Integrated Cascade Catalysts for Electrochemical Nitrate Reduction to Ammonia
https://doi.org/10.1021/jacs.5c03265利用可再生能源驱动的电催化硝酸盐还原反应(NO₃⁻-RR)为实现零碳排放氨(NH₃)合成提供了一条极具前景的途径。然而,缓慢的质子-电子耦合转移及副产物生成问题阻碍了高效氨合成。本研究构建了一种集成级联催化体系,阐明了硝酸盐还原反应过程中活性氢(H)生成与利用的主导机制。以原子级分散的铁(Fe)位点锚定于氮掺杂碳基质并包覆钌(Ru)纳米颗粒的代表性催化剂为例,该催化剂在相对于可逆氢电极(RHE)0 V的低电位下,氨产率高达2336.43 μgNH₃ h⁻¹ mgₐₜ⁻¹,同时法拉第效率达96.03%。此外,原位同步辐射傅里叶变换红外光谱(SR-FTIR)与密度泛函理论(DFT)计算表明,电子从铁原子向钌颗粒的转移不仅增强了铁位点对氮氧化物(NOₓ⁻)物种的亲和力,还提高了钌位点的氢覆盖度,从而加速加氢步骤并维持稳定的H生成-消耗循环。本研究揭示了级联催化结构中活性氢的机制起源,为推进高选择性、高能效及持久耐用的氨电合成及废水处理提供了基础性见解。氨(NH₃)作为现代社会最重要的基础化学品之一,因其高能量密度,既是不可或缺的工业原料,也是极具潜力的无碳能源载体。目前,高能耗的哈伯-博世(H-B)工艺仍是大规模合成氨的主要工业途径,但其存在能耗高、碳排放量大及对化石燃料依赖等问题。以水为质子源的可再生能源驱动型氨合成路线,因其温和的反应条件、环境友好性和高理论效率而备受关注。然而,氮氮三键(N≡N)的高解离能(941 kJ mol⁻¹)和氮气(N₂)的低溶解度成为制约氨产率提升的主要障碍,其产率较H-B工艺低两个数量级。为弥合这一差距,深入理解氮循环促使研究聚焦于含氮活性物种(如一氧化氮和硝酸盐)的回收再利用以合成氨。硝酸盐(NO₃⁻)具有高溶解度和低氮氧双键(N═O)解离能(204 kJ mol⁻¹)的特点,有助于降低电解过程中的传质限制和能耗。同时,硝酸盐广泛分布于工业废水和液态核废料中,对人类健康和水生生态系统构成严重威胁。因此,在温和反应条件下通过电化学途径将废弃硝酸盐转化为高附加值氨,对于缓解环境负担并创造经济效益具有重要现实意义。硝酸盐还原反应(NO₃⁻-RR)涉及复杂的质子耦合电子转移过程,可通过5电子(生成N₂)或8电子(生成NH₃)路径选择性进行。反应过程中存在多种稳定性及吸附能力各异的中间体,包括氨、亚硝酸盐、联氨、羟胺、一氧化氮和一氧化二氮,其氧化态范围从-3至+5,进一步增加了特定电极表面反应机制的理解难度。直接反应机制始于硝酸根离子在电催化剂表面的吸附,随后通过质子耦合电子转移或吸附原子氢介导的路径进一步还原为NO₂⁻。然而,由于硝酸根的对称共振结构及其与水分子间的强氢键作用,电子向NO₃⁻最低未占据分子轨道(LUMO)的转移需克服高能垒,因此需采用适宜电极以加速反应。既往研究主要聚焦于过渡金属,其中部分具有未配对d电子且d带能量与硝酸根π* LUMO相近的金属(如铜、银和铂)可能对硝酸盐还原具有电活性。然而,裸铜催化剂在NO₃⁻-RR过程中易因对含氮中间体的强吸附能力而迅速失活。近期,通过调控铜中心吸附的部分还原中间体的质子/电子转移动力学及结合强度,显著缓解了上述限制,具体方法包括铜与贵金属或其他过渡金属(如铂、钯和镍)的合金化,或与分子固体载体(如氮化硼碳、MXenes)或金属氧化物(如氧化亚铜、氧化钴)形成杂化物。然而,受限于标度关系,这些进展通常需要高浓度硝酸盐(如1 M)或相对负的起始电位(<−0.2 V vs RHE)以平衡NO₃⁻至NO₂⁻和NO₂⁻至NH₃的反应速率,导致能耗增加。此外,鉴于亚硝酸盐中间体(NO₂)在电极表面的高反应性,后续加氢步骤对产物分布具有关键控制作用。一方面,若NO₂结合较弱,可能从电极表面脱附,导致不期望的NO₂⁻积累,降低氨的法拉第效率(FE)并给实际水处理带来挑战。另一方面,若NO₂⁻稳定吸附,可进一步电化学还原为NO,而NO后续步骤的速率差异将决定最终产物分布。因此,为提高氨选择性,需确保充足的质子源供应,以促进含氮中间体经不同氧化态连续加氢生成氨,该过程在很大程度上决定了水分解中的Volmer步骤。然而,NO₃与H吸附不匹配导致单一活性位点催化剂仅能在更负电位下同时达到足够的NO₃与H覆盖度。因此,涉及多中间体和多步骤的多电子转移反应凸显了设计功能特异性活性位点以优化各自反应路径的重要性。另一需考虑的问题是,若H的生成与消耗不匹配,或在对应位点加氢过程中易脱附,则H-H复合可能轻易发生,释放氢气并降低氨生产的法拉第效率。例如,张等人在应变钌纳米团簇上实现了1.17 mmol h⁻¹ cm⁻²的高氨生成速率,但由于竞争性析氢反应(HER),高法拉第效率仅能在狭窄电位窗口内维持。这凸显了在NO₃⁻-RR过程中,不同功能位点需紧密空间接近以促进氮或氢相关物种的有效传输,实现水分解提供充足质子供应与反应中间体及时消耗之间的平衡。此外,催化剂应具备一定的H储存能力,同时确保优异的水分解性能,这表明传统随机物理混合物在调控理想反应序列方面存在局限性。受上述见解启发,本研究报道了一种通过在单原子基底上直接沉积纳米颗粒构建集成催化体系的尝试,以解决含氮物种H供应与积累的挑战。在该集成级联电催化剂中,钌纳米颗粒与原子级铁-氮₄(Fe-N₄)位点间的强相互作用促进了电荷重新分布,从而降低了反应能垒。在此催化剂构型中,钌物种可有效将H传递至对NO₃⁻及其衍生物具有高吸附亲和力的相邻铁位点。因此,优化后的RuNP@FeSA-N-C在相对于可逆氢电极(RHE)0 V的低电位下实现了高达96.03%的最大氨法拉第效率,同时在-0.4 V vs RHE下达到了具有工业相关性的氨产率(14837.65 μg h⁻¹ mgₐₜ⁻¹),超越了大多数最先进的NO₃⁻-RR催化剂。通过同位素竞争实验、原位同步辐射傅里叶变换红外光谱(SR-FTIR)和密度泛函理论(DFT)计算对反应机制的深入洞察进一步证实,铁位点负责NO₃⁻的吸附、脱氧和加氢,而邻近的钌位点则提供表面氢化过程所需的活化*H物种,以促进H-N键的形成。这种协同级联催化过程实现了NO₃⁻向NH₃的高效电还原。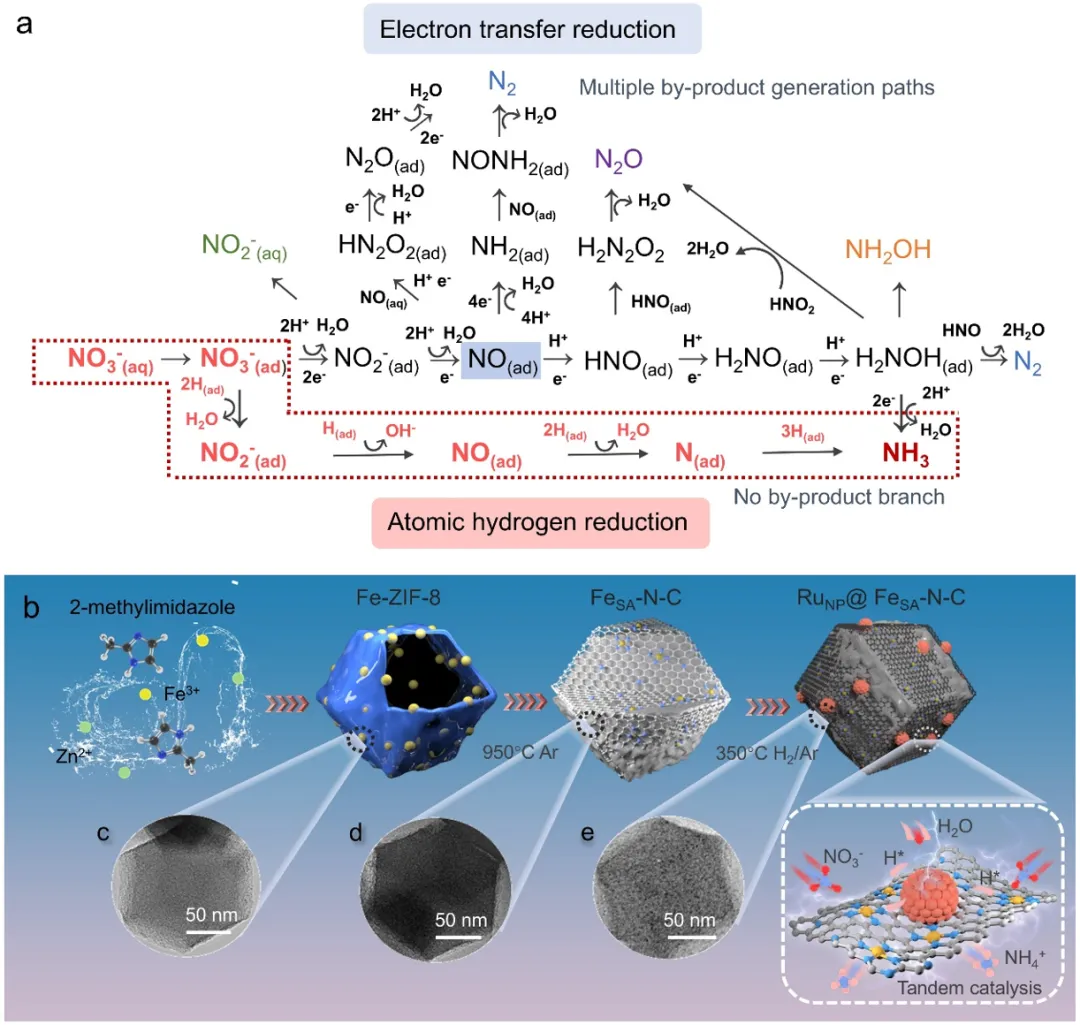
图1. 电化学硝酸盐还原反应催化剂的设计原则与合成。(a)水中硝酸盐电化学还原过程中的机理和主要过程示意图。(b)RuNP@FeSA–N–C合成过程示意图。(c)铁-沸石咪唑酯骨架结构材料-8(Fe-ZIF-8)、(d)FeSA–N–C和(e)RuNP@FeSA–N–C的TEM图像。

图2. RuNP@FeSA–N–C的形貌表征。(a)扫描电子显微镜(SEM)图像。(b)透射电子显微镜(TEM)图像及放大图像(插图)。(c)高分辨率透射电子显微镜(HRTEM)图像。(d)球差校正高角环形暗场扫描透射电子显微镜(HAADF-STEM)图像(虚线水黄色圆圈表示Ru纳米颗粒,红色圆圈表示铁单原子(Fe SAs))。(e)带有颗粒直径直方图的HAADF-STEM图像,以及(f)Ru、Fe、N和C的相应元素分布图。
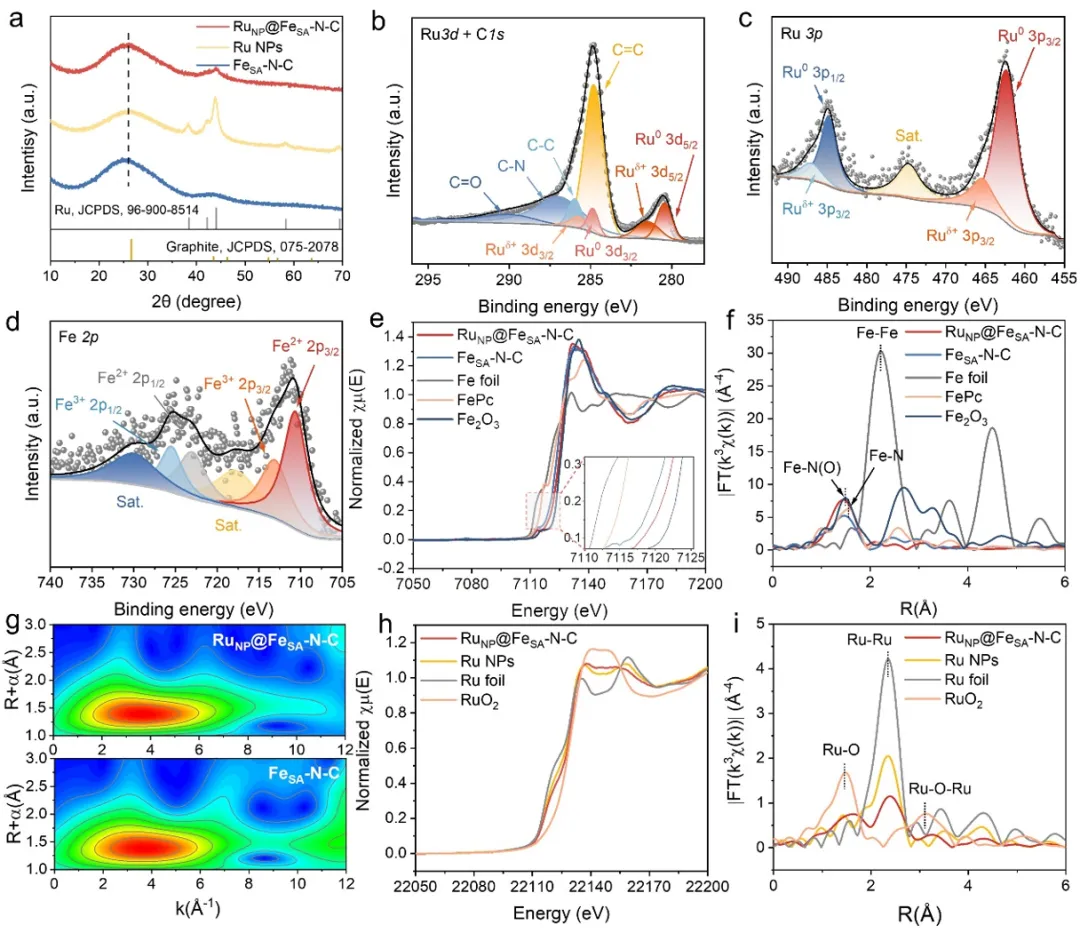
图3. 催化剂的原子结构分析。(a)X射线衍射(XRD)图谱。(b)制备样品中Ru 3d、(c)Ru 3p和(d)Fe 2p的高分辨率X射线光电子能谱(XPS)图谱。(e)归一化的Fe K边和(f)RuNP@FeSA–N–C、FeSA–N–C、三氧化二铁(Fe₂O₃)、酞菁铁(FePc)和铁箔在R空间的傅里叶变换扩展X射线吸收精细结构(FT-EXAFS)曲线。(g)RuNP@FeSA–N–C和FeSA–N–C的EXAFS信号的小波变换图。(h)RuNP@FeSA–N–C、Ru NPs、二氧化钌(RuO₂)和Ru箔的Ru K边X射线吸收近边结构(XANES)图谱,以及(i)在R空间的FT-EXAFS图谱。
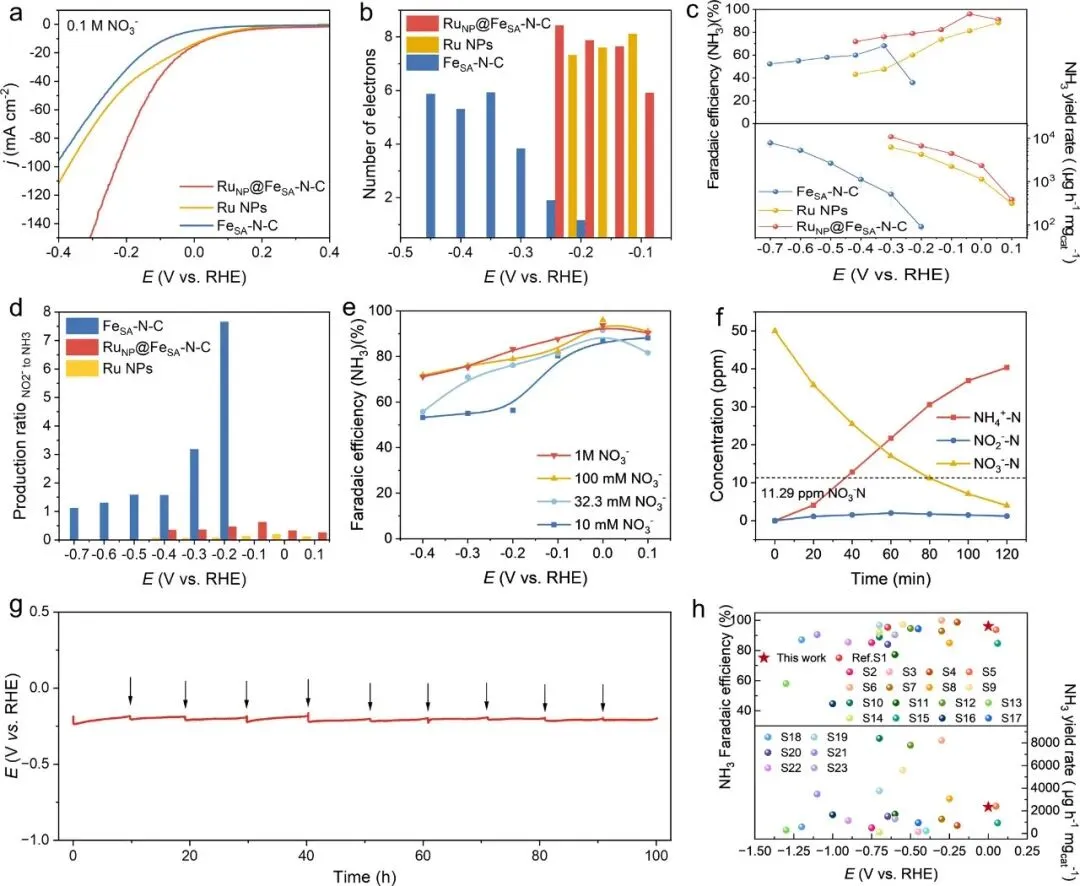
图4. NO₃⁻RR的电催化性能。(a)含100 mM KNO₃的1 M KOH溶液中的线性扫描伏安(LSV)曲线。(b)由Koutecky–Levich图得到的电子转移数与施加电位的关系。(c)RuNP@FeSA–N-C、Ru NPs和FeSA–N–C的电位依赖性NH₃法拉第效率和质量归一化产率。(d)在100 mM KNO₃ + 1 M KOH电解液中,相对于RHE电位为0 V时,不同样品组分生成NO₂⁻与NH₃的比率。(e)不同NO₃⁻浓度下,RuNP@FeSA–N–C在1 M KOH电解液中的NH₃法拉第效率。(f)相对于RHE电位为0 V时,RuNP@FeSA–N–C的NO₃⁻、NO₂⁻和NH₃浓度随时间的变化(虚线表示世界卫生组织(WHO)对饮用水设定的限值)。(g)在H型电池中,100 mA cm⁻²电流密度下,RuNP@FeA–N-C在0.1 M KNO₃ + 0.1 M KOH电解液中的长期电催化稳定性测试(黑色箭头表示更换新鲜电解液)。(h)RuNP@FeSA–N–C与其他广泛报道的电催化剂的电催化NO₃⁻RR性能比较。
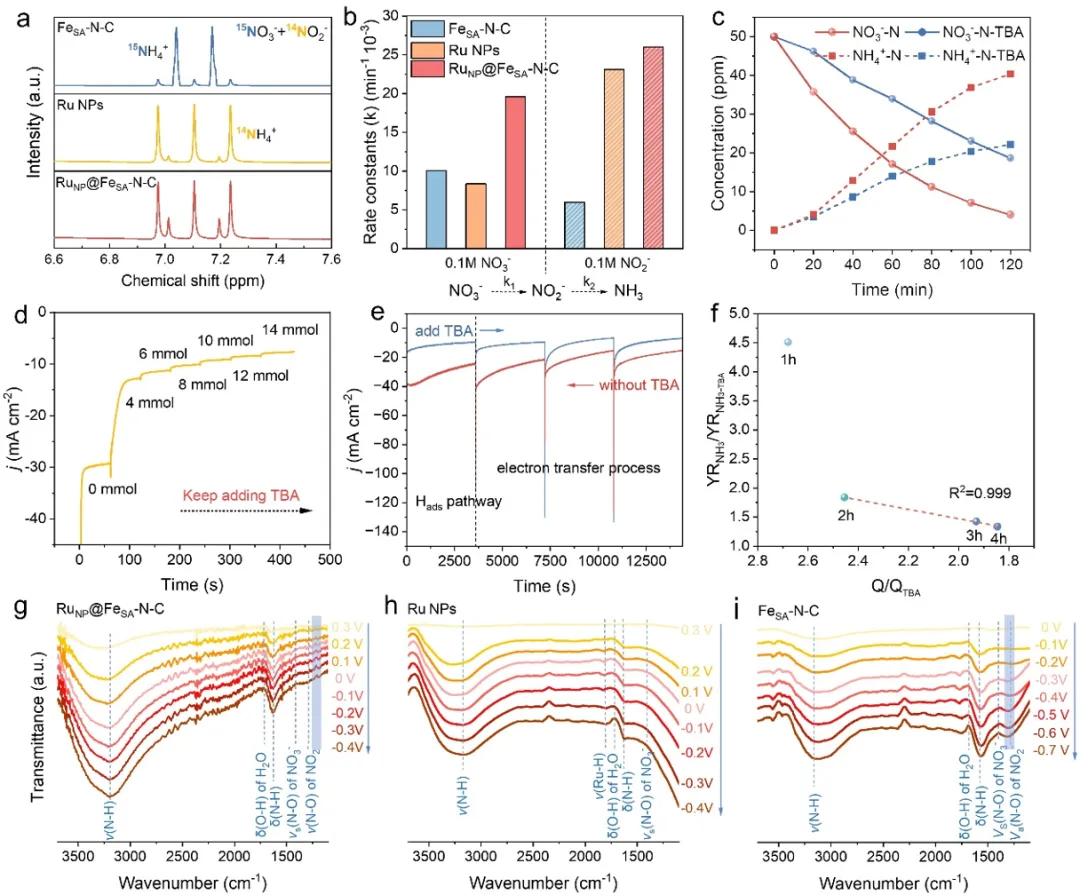
图5. NO₃⁻RR过程中催化剂的反应动力学、反应活性和选择性评估。(a)电解含100 mM ¹⁵NO₃⁻和100 mM ¹⁴NO₂⁻的1 M氢氧化钾(KOH)混合溶液后,电解液同位素竞争实验的高分辨率¹H核磁共振(NMR)谱图(600 MHz)。(b)RuNP@FeSA–N–C、Ru NPs和FeSA–N–C催化剂上NO₃⁻和NO₂⁻还原反应的计算反应常数(NO₃⁻转化为NO₂⁻的k₁和NO₂⁻转化为氨(NH₃)的k₂)。(c)有无10 mmol四丁基铵(TBA)时NO₃⁻还原和NH₃生成的浓度变化。(d)添加不同剂量TBA时,RuNP@FeSA–N–C在NO₃⁻RR过程中的计时安培曲线。(e)在相对于可逆氢电极(RHE)电位为0 V时,有无TBA存在下的NO₃⁻RR的电流-时间(I-t)曲线,以及(f)NH₃生成比率与电荷比率的关系。在氩气饱和的1 M KOH + 0.1 M硝酸钾(KNO₃)电解液中,NO₃⁻RR过程中不同施加电位下,(g)RuNP@FeSA–N–C、(h)Ru NPs和(i)FeSA–N-C的原位同步辐射傅里叶变换红外光谱(SR-FTIR)图谱。
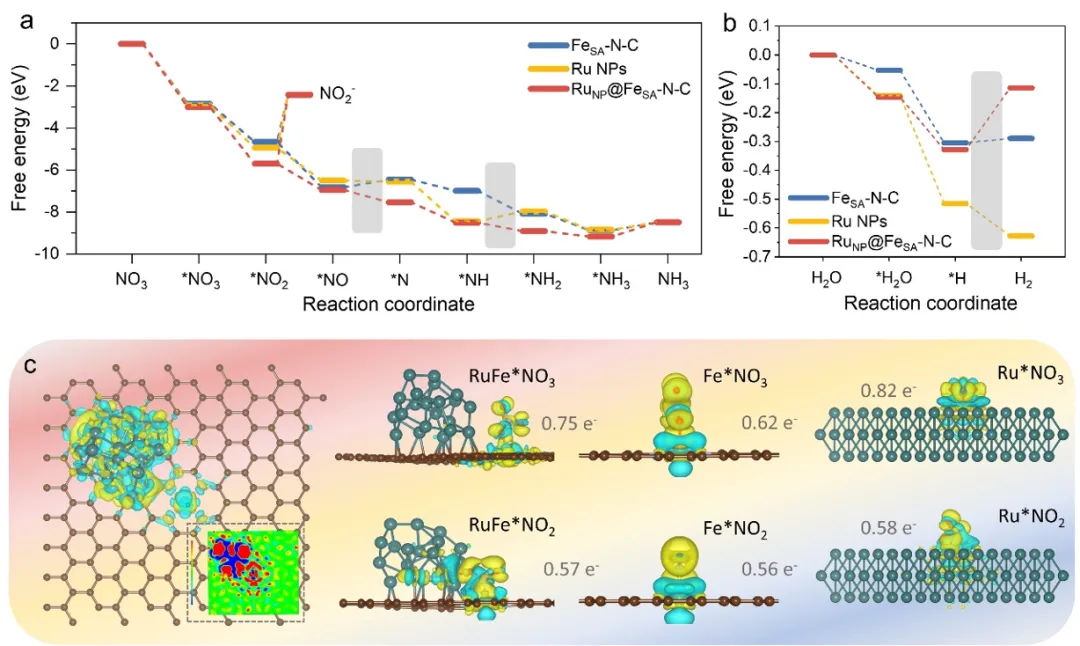
图6. 密度泛函理论(DFT)计算。(a)NO₃⁻还原反应(NO₃⁻RR)和(b)析氢反应(HER)过程中,在RuNP@FeSA–N-C、Ru纳米颗粒(Ru NPs)和FeSA–N–C表面生成的不同中间产物的反应自由能。(c)RuNP@FeSA–N-C界面的差分电荷密度,以及RuNP@FeSA–N–C、Ru NPs和FeSA–N–C吸附NO₃⁻和NO₂⁻后的电荷转移情况。黄色和蓝色等高线分别代表电子密度积累和耗尽。
综上所述,我们提出了一种以合理设计原则为指导的有效方法,用于开发硝酸盐还原制氨的协同催化体系。得益于优化的H吸附性能以及NO₃⁻还原反应(NO₃⁻RR)路径中中间产物的有利吸附行为,所开发的RuNP@FeSA–N-C在相对于可逆氢电极(RHE)电位为0 V的条件下,展现出高达2336.43 μgNH₃ h⁻¹ mgcat⁻¹的氨产率,同时对氨的法拉第效率(FE)高达96.03%,且在20次循环后性能下降可忽略不计。多种表征分析结合密度泛函理论(DFT)计算表明,H的生成及其被含氮中间产物及时消耗,对于同时提高NO₃⁻RR的法拉第制氨效率(FENH₃)和产率至关重要。通过调控电子结构,两个活性位点之间的协同效应不仅降低了NO₃⁻RR的能垒,还提高了深度加氢能力。同时,与Ru纳米颗粒催化剂相比,该协同效应避免了活性位点上H的过度占据,而后者会影响氨的法拉第效率。这种构建协同催化位点的策略,为高效生成和可控利用H提供了更深入的见解,可促进NO₃⁻RR过程中氮中间产物的加氢反应。此外,该策略还支持涉及多电子和多步反应的复杂电催化过程,对高能效的工业过程具有潜在益处。








 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
