中国科学技术大学傅尧/陆熹&南京农业大学卢倩倩最新Angew丨Ni/Co催化环己烷立体发散酰胺化合成
- 2026-06-21 12:06:04


三维有机分子结构的构建是现代有机化学的核心课题。四面体碳构型理论建立了以sp3杂化碳为中心的立体化学框架,推动分子空间结构研究从“平面认知”走向“三维探索”。对于二取代环己烷,其以顺式和反式两种构型存在,取代基的取向决定了热力学稳定性,并导致物理化学性质和生物活性的显著差异。然而,饱和环烷烃取代基的精确立体控制仍然具有挑战性。
外环不饱和键的还原是多取代环烷烃立体发散性合成的经典案例,其中取代环己酮/环状亚胺的还原已被广泛研究。其极性C=O/C=N键易于与金属氢化物反应,立体选择性受复杂而巧妙的立体和电子效应控制。相比之下,外环C=C键的立体控制则较为有限:氢化物与非极性C=C键之间的极性不匹配阻碍了直接加成,且轴向/赤道攻击的差异不明显,使得选择性控制更为复杂。
过渡金属催化体系为解决这一挑战提供了可能的方案。通过合理调控金属或配体,可以精确调控金属氢化物物种或金属-官能团物种的空间结构和电子性质。这使得活性物种能够高选择性地加成到碳碳双键上,从而实现非对映发散性合成。
然而,过渡金属的引入可能伴随烯烃异构化、无效的原脱金属以及官能化试剂的分解等副反应。另一方面,反应机理的复杂性增加了选择性控制的不确定性。因此,过渡金属催化的外环C=C键氢官能化反应为多取代环烷烃的非对映发散性合成带来了机遇与挑战。
在此,研究者报道了亚甲基环己烷的非对映发散性氢酰胺化反应。具体而言,镍催化体系能够高效合成1,4-顺、1,4-反、1,3-顺、1,3-反和1,2-顺二取代环己烷,而钴催化体系则实现了1,2-顺和1,2-反的立体发散性合成。在机理上,成功控制反应选择性的关键在于差异化活性催化物种策略,该策略精确调控了金属氢化物或金属-官能团物种的空间结构和电子性质。
该研究拓宽了多取代环己烷合成的化学空间,为含环烷基药物分子的合成提供了有力工具,并为有机化学中“取代环己烷立体化学”的研究提供了重要案例。
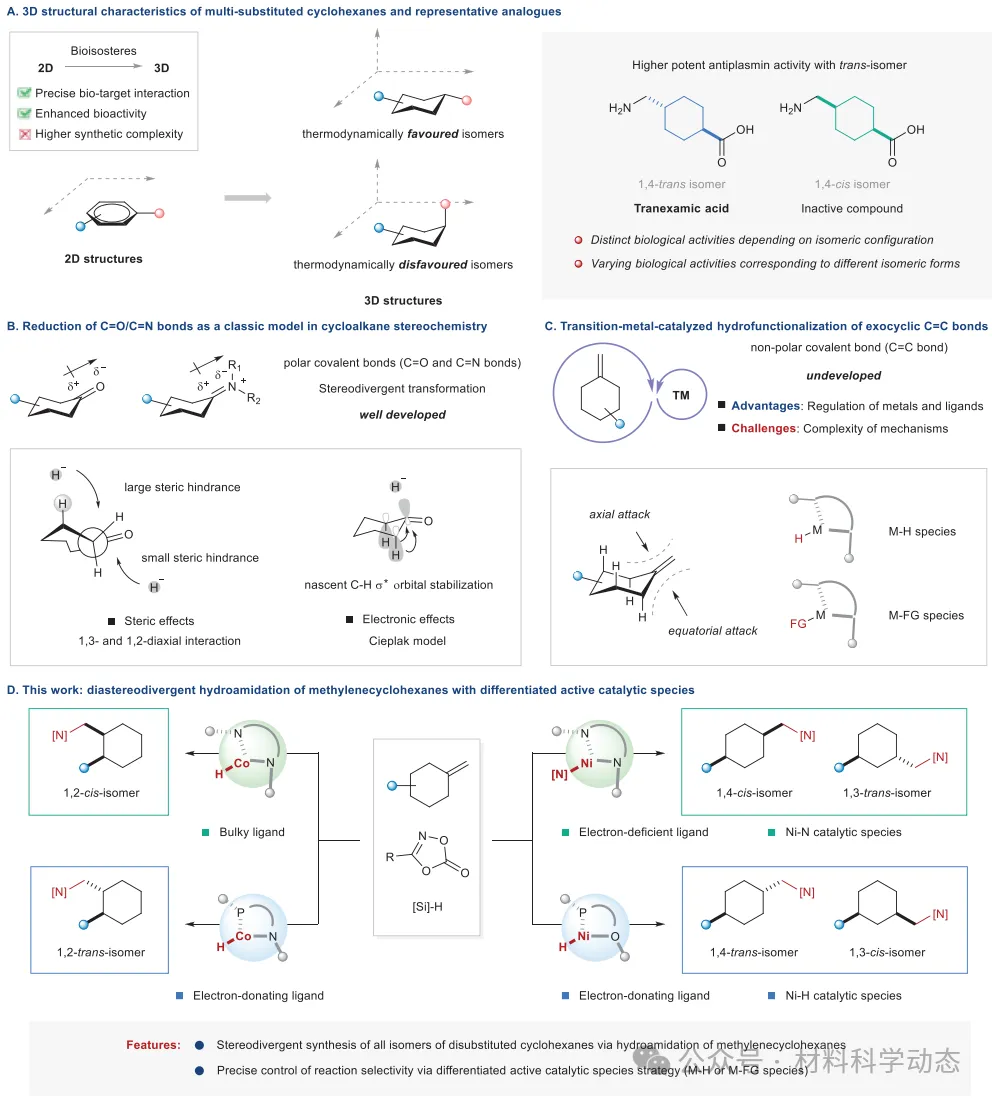
图1:多取代环烷烃的合成。 M,金属;FG,官能团;[N],酰胺基或其他含氮官能团。
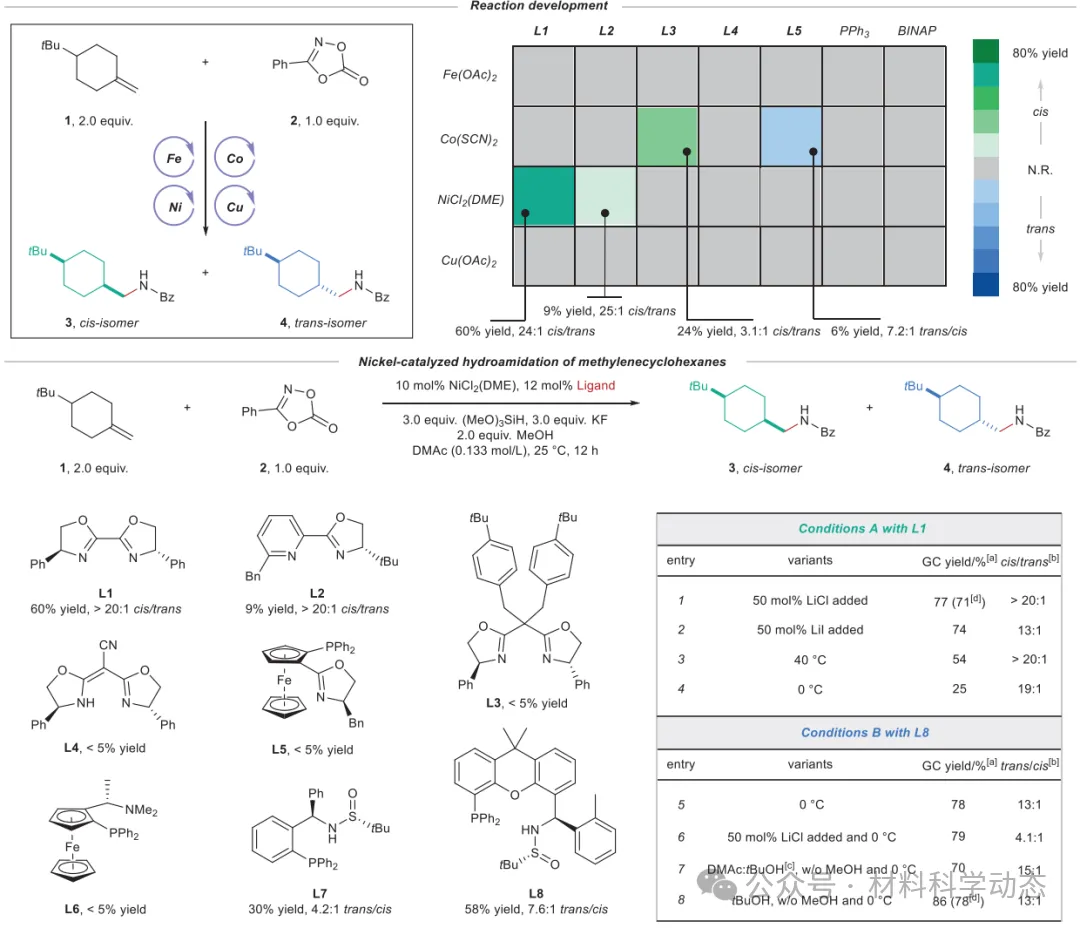
图2:亚甲基环己烷非对映发散性氢酰胺化反应的条件优化。 反应在氩气气氛下进行。条件:1(0.20 mmol,2.0 equiv)、2(0.10 mmol,1.0 equiv)、金属催化剂(10 mol%)、配体(12 mol%)、硅烷(3.0 equiv)、碱(3.0 equiv)、醇(2.0 equiv)、溶剂(0.133 mol/L)、25°C、12 h,0.1 mmol规模。[a] GC产率,以三苯基甲烷为内标。[b] 面选择性由GC分析确定。[c] DMAc:tBuOH(v:v = 4:1)。[d] 括号内为分离产率。tBu,叔丁基;Bz,苯甲酰基;OAc,乙酰氧基;SCN,硫氰酸根;DME,1,2-二甲氧基乙烷;BINAP,2,2'-双(二苯基膦基)-1,1'-联萘;DMAc,N,N-二甲基乙酰胺;Bn,苄基。
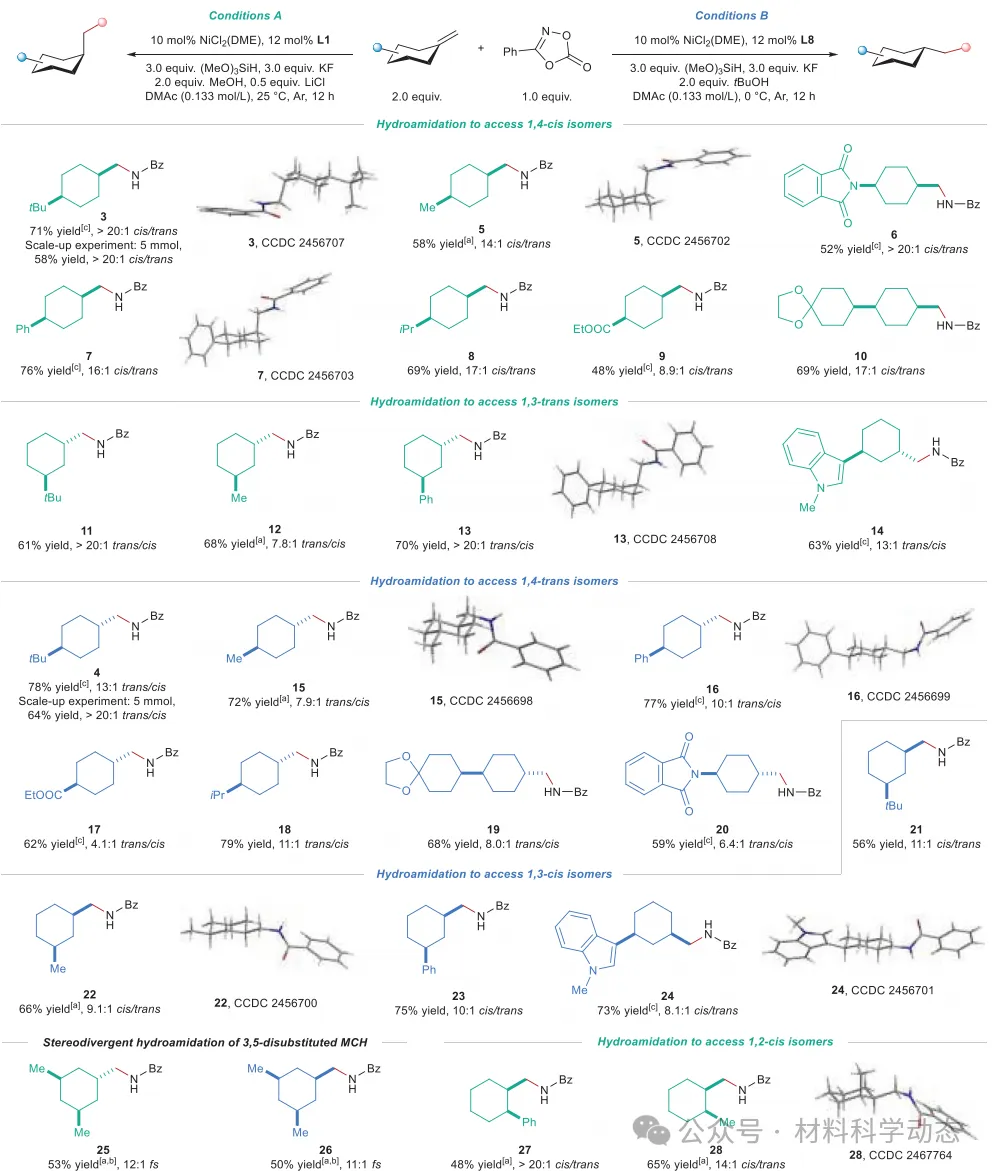
图3:亚甲基环己烷的底物拓展。 条件A和条件B的反应均在0.20 mmol规模下进行,分离产率,面选择性由GC分析确定。[a] 使用3.0当量烯烃。[b] 面选择性由1H NMR谱分析确定。[c] 两种异构体可通过柱色谱分离。fs,面选择性。
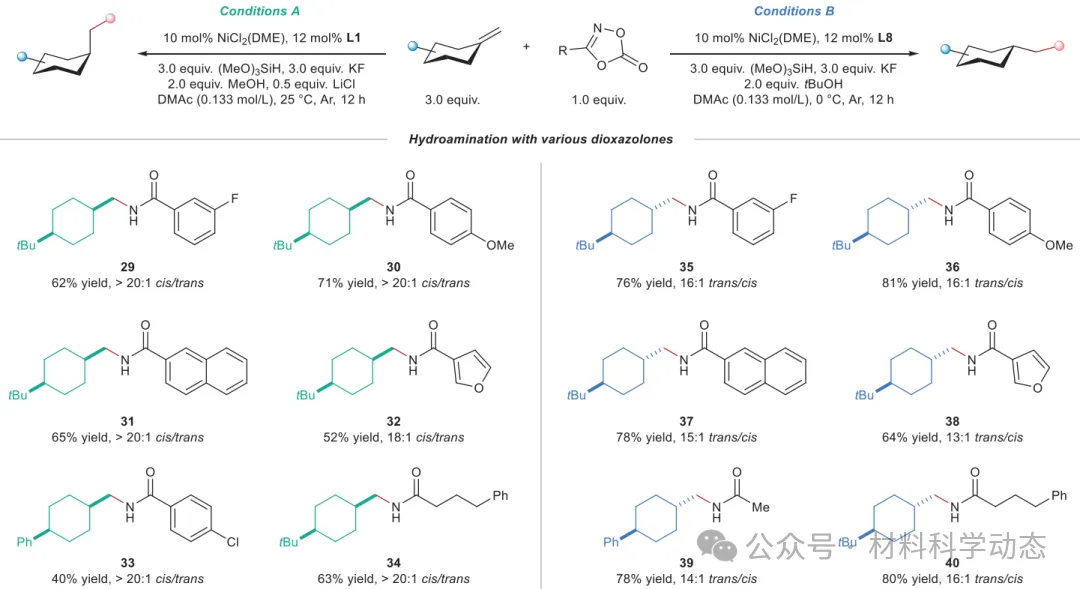
图4:二噁唑酮的底物拓展。 条件A和条件B的反应均在0.20 mmol规模下进行,分离产率,面选择性由GC分析确定。
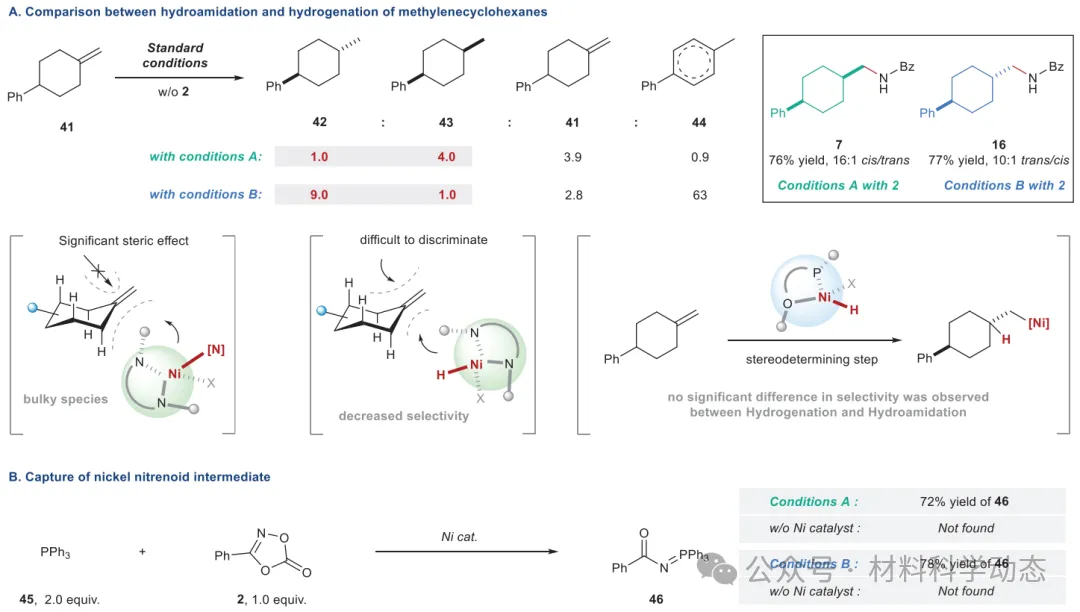
图5:反应机理探究。 条件A和条件B的反应均在0.20 mmol规模下进行,分离产率,面选择性由GC分析确定。
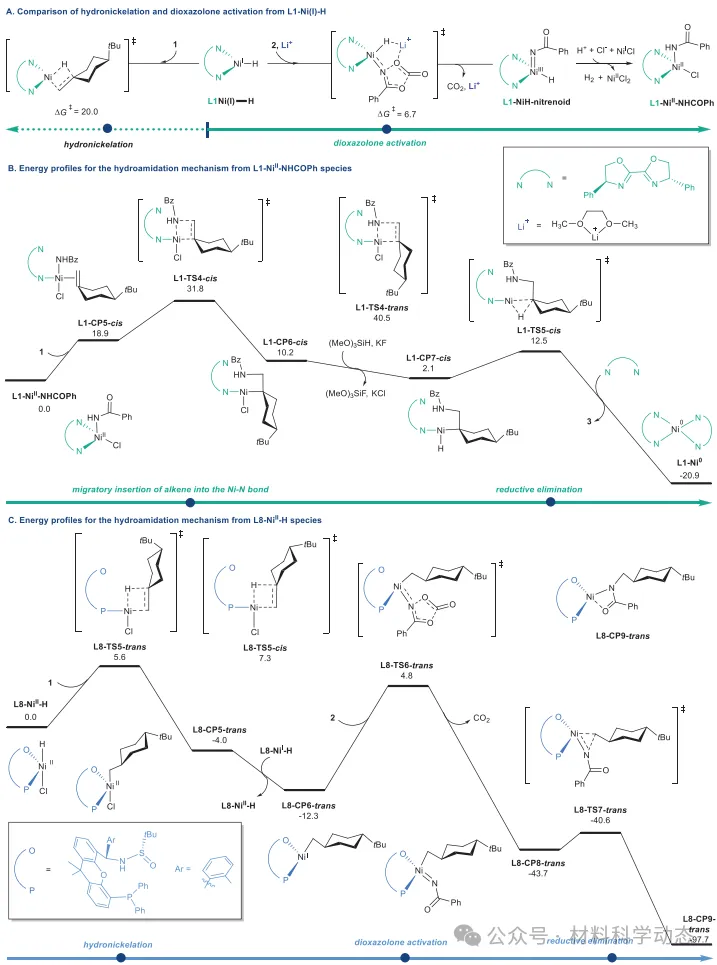
图6:反应路径的DFT计算。 吉布斯自由能计算采用M06/6-311+G(d,p)-SDD/SMD//B3LYP/D3(BJ)/6-31G(d)-SDD理论水平,能量单位为kcal/mol。
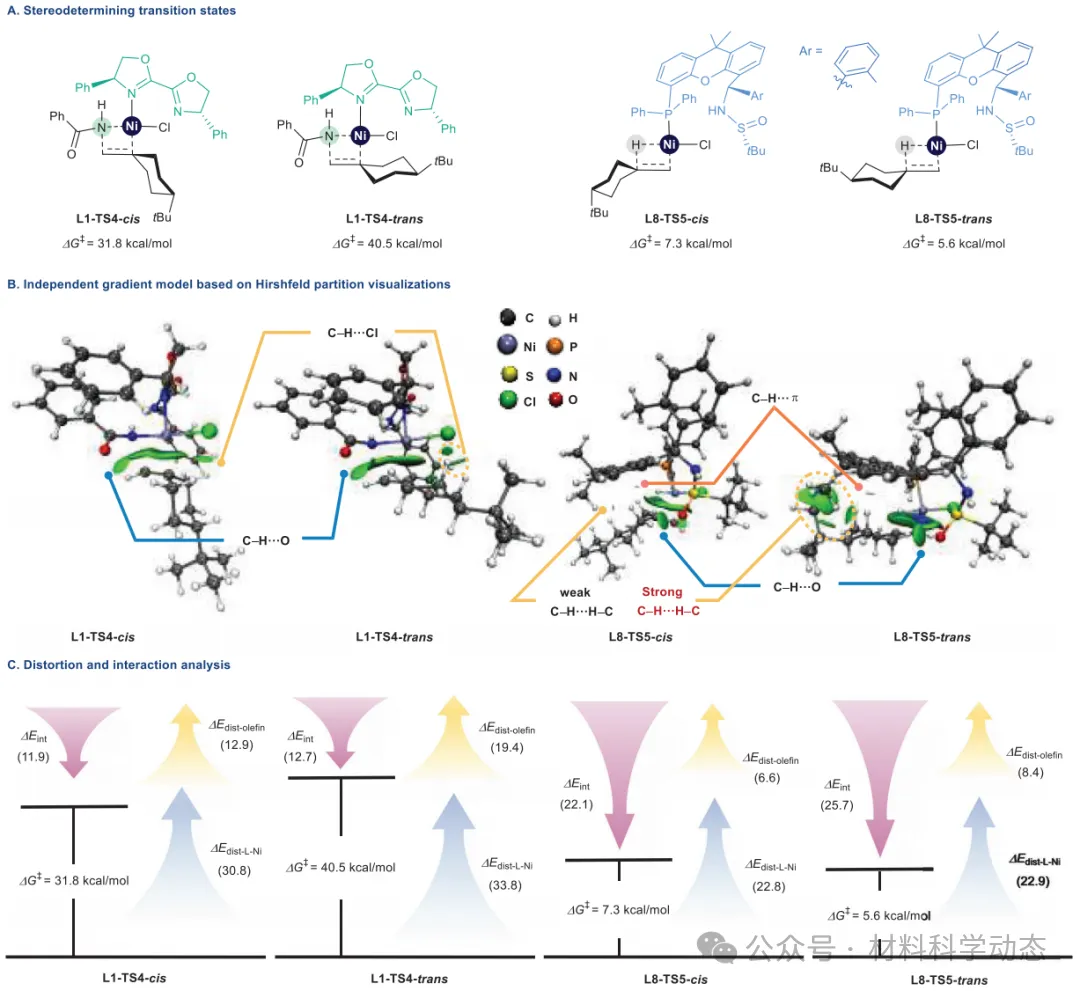
图7:选择性决定过渡态的分析。 吉布斯自由能计算采用M06/6-311+G(d,p)-SDD/SMD//B3LYP/D3(BJ)/6-31G(d)-SDD理论水平,能量单位为kcal/mol。
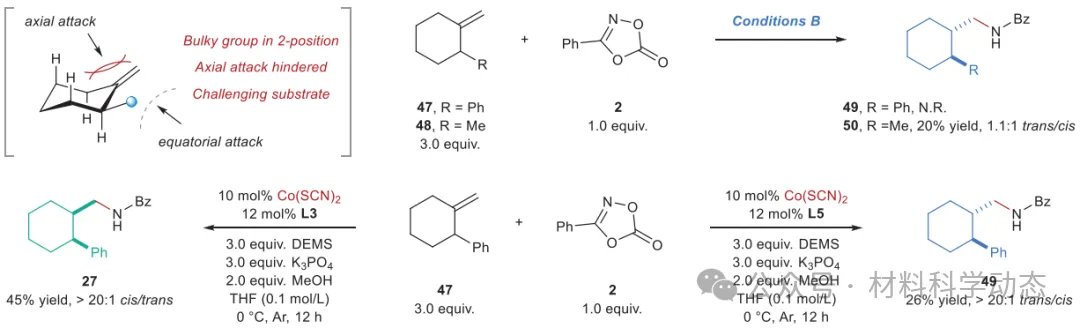
图8:钴催化立体发散性氢酰胺化合成1,2-异构体。 反应在0.20 mmol规模下进行,分离产率,面选择性由GC分析确定。N.R.,无反应;DEMS,1,2-二甲氧基乙烷。
该研究报道了亚甲基环己烷的非对映发散性氢酰胺化反应,通过镍和钴两种催化体系实现了二取代环己烷所有构型的立体选择性合成。镍催化体系高效合成1,4-顺/反和1,3-顺/反异构体,钴催化体系则实现1,2-顺/反的立体发散性合成,共获得43个产物,产率最高达86%,选择性最高>20:1。
机理研究和DFT计算表明,关键在于差异化活性催化物种策略。L1/Ni体系通过Ni-N物种对烯烃的插入,利用立体位阻效应实现热力学不利的顺式产物;L8/Ni体系则通过Ni-H物种对烯烃的插入,借助C-H···H-C弱相互作用实现热力学有利的反式产物。钴催化体系通过Co-H物种对烯烃的插入,补充了1,2-异构体的合成。
配体筛选使用了125种配体,发现BiOX类配体有利于顺式产物,SadPhos类配体则实现选择性反转。该工作拓宽了多取代环己烷的化学空间,为含环烷基药物分子的合成提供了有力工具,并为取代环己烷立体化学研究提供了重要案例。
Diastereodivergent Hydroamidation of Methylenecyclohexanes With Differentiated Active Catalytic Species,Angewandte Chemie International Edition,2026,https://doi.org/10.1002/anie.9587119
#有机合成 #非对映发散性合成#氢酰胺化#环己烷#镍催化#钴催化#中国科学技术大学#南京农业大学#Angew#顶刊解读


