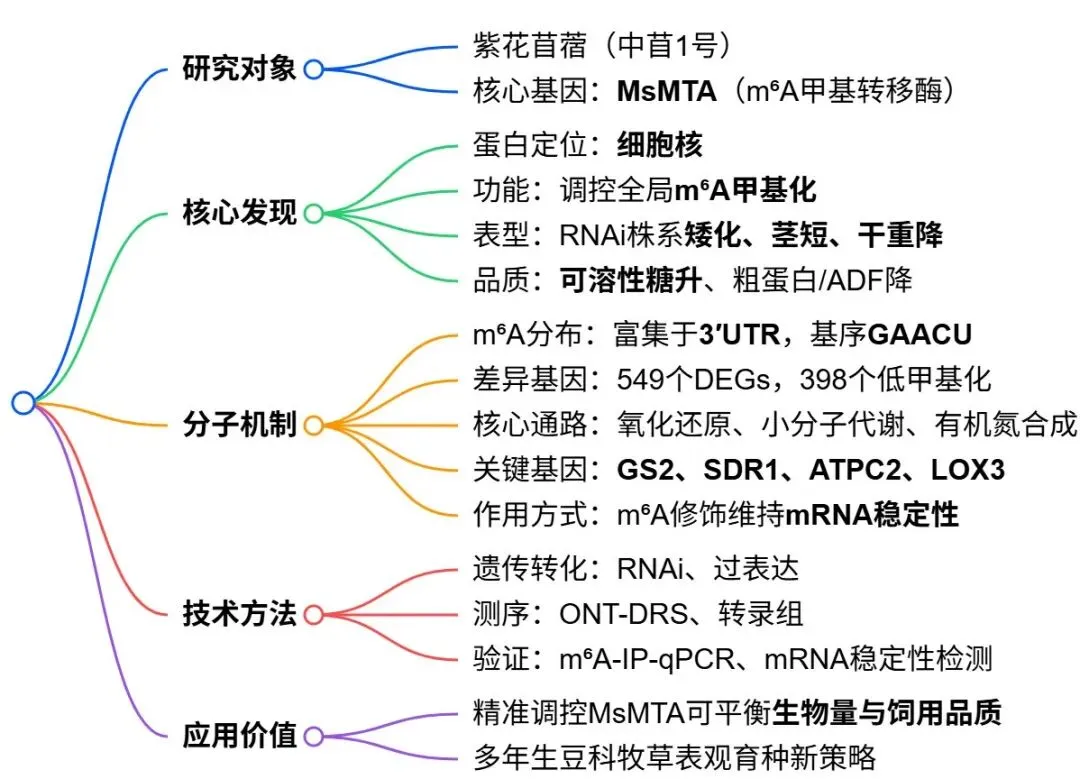
HR(Q1/8.5)|中国农业大学张蕴薇教授团队揭示紫花苜蓿m⁶A甲基转移酶MsMTA调控碳氮代谢平衡的分子机制
- 2026-05-09 07:26:41




研究背景
N⁶-甲基腺苷(m⁶A)是真核生物mRNA中最丰富的内部修饰,其通过“书写蛋白”、“擦除蛋白”和“阅读蛋白”的协同作用,在转录后水平广泛调控RNA的剪接、输出、翻译及稳定性。在植物中,以MTA为核心的甲基转移酶复合体介导的m⁶A修饰在胚胎发生、器官形态建成以及非生物胁迫响应等生理发育过程中发挥着至关重要的作用。本研究以重要的多年生豆科牧草紫花苜蓿(Medicago sativa)为研究对象。紫花苜蓿具有极高的经济和生态价值,其茎秆的伸长与木质化程度、以及碳氮代谢的分配平衡,直接决定了该作物的生物量产量与最终的营养品质。
近年来,随着全转录组测序技术的发展,模式植物中的m⁶A甲基化图谱已被广泛解析,揭示了其在编码区和3′非翻译区(3′UTR)的富集特征及其对mRNA命运的决定性影响。然而,在多年生豆科作物领域,关于m⁶A甲基化的研究仍处于起步阶段。以往针对紫花苜蓿生长发育和营养代谢的研究多局限于传统的植物激素信号传导或DNA水平的转录调控,而极少涉及RNA表观修饰层面。迄今为止,紫花苜蓿中m⁶A甲基转移酶的核心基因组成、亚细胞定位及其在协调植物生长与碳氮代谢网络中的具体表观转录组学功能尚未得到系统性的鉴定与解析。
当前该领域面临的核心挑战在于如何精准解析表观转录组修饰与植物宏观农艺性状(如生物量和营养品质)之间的分子联系。本研究通过鉴定紫花苜蓿的核心m⁶A甲基转移酶基因MsMTA,揭示了其通过维持氧化还原、小分子代谢及有机氮合成相关基因(如GS2、SDR1等)转录本稳定性来调控代谢稳态的机制。这一发现首次在紫花苜蓿中证实了m⁶A修饰对关键代谢酶RNA命运的直接控制作用,不仅在理论上阐明了多年生豆科植物偶联生长发育与碳氮代谢的表观遗传机制,也为未来通过靶向操纵m⁶A甲基化途径、培育兼具高蛋白与高可溶性糖的优质高产紫花苜蓿新品种提供了重要的分子靶点和改良策略。
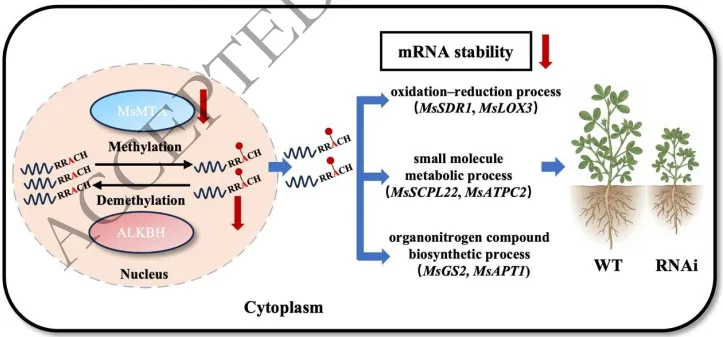
本研究首次在紫花苜蓿中鉴定出核定位的 m⁶A 甲基转移酶 MsMTA,证实其为调控全局 m⁶A 修饰的核心因子;沉默 MsMTA会使整体 m⁶A 水平下降43.41%–58.35%,引发植株矮化、茎秆伸长受阻,同时重塑碳氮分配,使可溶性糖显著升高、粗蛋白与酸性洗涤纤维小幅降低;机制上,MsMTA 通过 m⁶A 修饰稳定GS2、SDR1、ATPC2等代谢关键基因的 mRNA,调控氧化还原、小分子代谢与有机氮合成通路,为苜蓿产量与品质协同改良提供表观转录组新靶点。
研究方法
2.1 试验材料与表型鉴定
为探究目标基因对紫花苜蓿生长发育的影响,本研究以‘中苜1号’紫花苜蓿(Medicago sativa)为试验材料,在受控的人工气候室条件下进行标准化培养。通过扩增MsMTA基因的全长编码序列,分别构建了由CaMV 35S启动子驱动的过表达载体和RNA干扰载体。借助根癌农杆菌(Agrobacterium tumefaciens)介导的叶片外植体转化法,成功获得了野生型(WT)及转基因株系。在植株移栽后的第5至10周期间,对各株系(均设置五个生物学重复)的株高、节间长度以及茎粗等关键表型指标进行了系统的动态监测与定量评估,以明确基因功能对植物株型和生物量积累的具体影响。
2.2 分子克隆与多组学测序设计
为阐明MsMTA的亚细胞定位及全转录组m⁶A修饰图谱,开展了一系列分子与组学试验。首先,将MsMTA与绿色荧光蛋白(GFP)融合,通过农杆菌注射本氏烟草(Nicotiana benthamiana)叶片并在共聚焦显微镜下进行亚细胞定位观察。随后,提取高质量的总RNA并分离Poly(A)⁺ RNA,利用抗m⁶A抗体通过斑点杂交试验对整体m⁶A丰度进行定量。在组学测序方面,提取WT和MsMTA-RNAi植株的混合RNA样本,利用Oxford Nanopore Technologies (ONT) MinION平台进行直接RNA测序(ONT-DRS)。数据分析采用基于似然比评分的二项/多项分布模型来鉴定差异甲基化区域(设定阈值为Score > 3)。
2.3 基因表达、转录本稳定性与统计学分析
为验证高通量测序结果并深入解析m⁶A修饰对靶基因的调控机制,研究采用了m⁶A-IP-qPCR和实时荧光定量PCR(RT-qPCR)技术。在m⁶A-IP-qPCR中,利用特异性抗体富集m⁶A修饰的RNA片段,并通过2⁻ΔCt方法计算相对富集度;常规RT-qPCR则以MsActin为内参基因,采用2⁻ΔΔCt方法量化目标基因的相对表达水平。此外,通过施加50 μg/mL放线菌素D抑制新的RNA合成,并在0、3、6、9小时等多个时间点取样,精确评估了目标mRNA的降解速率与转录本稳定性。所有试验数据均导入SPSS 17.0软件进行统计学分析,采用单因素方差分析(ANOVA)结合Duncan多重极差检验评估组间差异,统计显著性阈值严格设定为P < 0.05。
研究结果
3.1 MsMTA是紫花苜蓿中m⁶A甲基化必需的核定位蛋白
序列与表达:MsMTA含有保守的MT-A70甲基转移酶结构域,在根、成熟叶和根瘤组织中具有较高的表达丰度。
亚细胞定位:烟草瞬时表达试验的荧光信号表明,MsMTA蛋白特异性定位于细胞核内。
功能验证:构建RNAi和过表达株系后,RNA斑点杂交证实MsMTA敲降会显著降低整体m⁶A水平,表明其为m⁶A沉积所必需。
MsMTA作为MTA/METTL3的同源物,具有典型的甲基转移酶结构域且定位于细胞核。通过构建并筛选出具有显著沉默效率的RNAi株系和高表达的过表达株系,进一步的RNA斑点杂交分析证实,抑制MsMTA表达会大幅度降低紫花苜蓿总RNA的m⁶A修饰丰度,确立了其在m⁶A甲基化过程中的核心功能地位。(Fig.1)
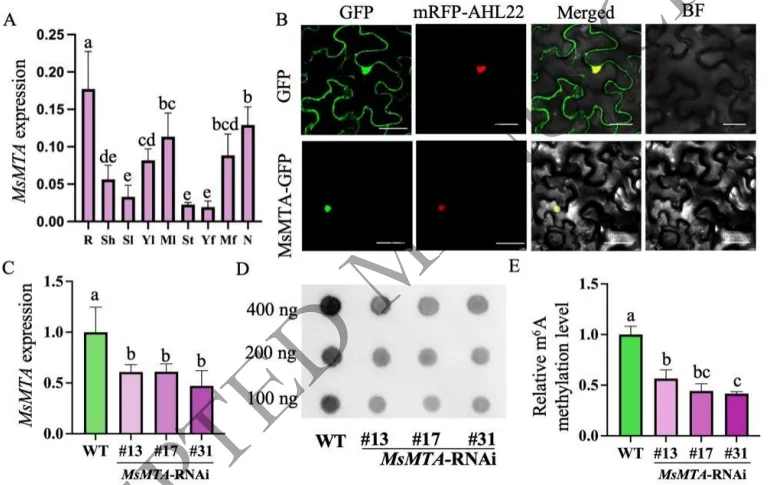
图1. MsMTA的功能分析。(A) MsMTA在紫花苜蓿不同组织中的表达模式。R,10天苗龄的根;Sh,10天苗龄的茎;Sl,10天苗龄的单叶;Yl,幼叶(折叠叶);Ml,成熟叶(展开叶);St,茎;Yf,幼花;Mf,成熟花;N,根瘤。(B) MsMTA的亚细胞定位。35S:GFP作为阳性对照。GFP:绿色荧光蛋白。比例尺=50 μm。(C) MsMTA-RNAi紫花苜蓿株系中MsMTA的相对表达水平。(D) MsMTA-RNAi紫花苜蓿株系总RNA m⁶A修饰水平的检测。(E) 使用ImageJ测量相对光密度,对(D)中斑点杂交的m⁶A修饰进行定量。
3.2 关键m⁶A甲基转移酶MsMTA调控紫花苜蓿的生长发育与营养品质
生长受抑:MsMTA沉默导致植株显著矮化,表现为节间缩短、茎变细及干物质积累减少,但叶片发育无显著差异。
营养改变:RNAi株系呈现出可溶性糖显著增加、粗蛋白和酸性洗涤纤维适度降低的“高糖、低蛋白、低纤维”特征。
过表达表型:过表达株系在株高、茎粗、节间长度及各项营养品质指标上与野生型相比均无显著差异。
抑制MsMTA表达主要限制了紫花苜蓿茎的伸长与增粗,导致植株呈现明显的矮化表型及干物质积累减少。同时,该基因的沉默重塑了碳氮营养分配,使得植株形成可溶性糖升高而粗蛋白和酸性洗涤纤维降低的特定营养特征。这些结果表明MsMTA不仅调控茎的发育,还深刻影响碳氮代谢与细胞壁成分的沉积,而其过表达则不足以引起显著的表型改变。(Fig.2)
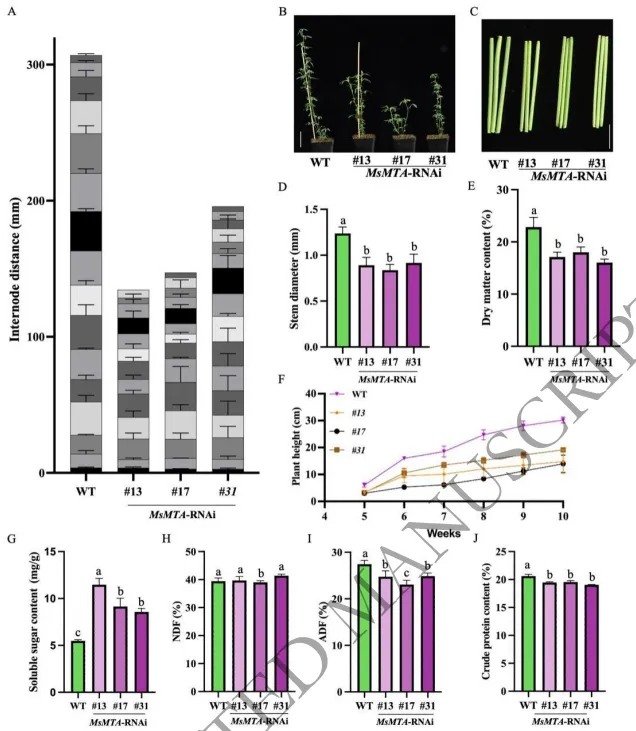
图2. 沉默MsMTA限制紫花苜蓿茎生长并改变营养组成。(A) 10周龄野生型(WT)植株与三个独立MsMTA-RNAi株系(#13、#17和#31)的节间长度比较。误差线表示至少五个生物学重复的标准差(SD)。(B) WT和MsMTA-RNAi紫花苜蓿植株的生长表型。比例尺=5 cm。(C) WT和MsMTA-RNAi株系茎形态的比较。比例尺=5 mm。(D) WT和MsMTA-RNAi植株茎粗的定量。(E) WT和MsMTA-RNAi植株干物质含量的测定。(F) 显示WT和MsMTA-RNAi株系10周内株高的生长曲线。(G) WT和MsMTA-RNAi株系可溶性糖含量的测定。(H) WT和MsMTA-RNAi株系中性洗涤纤维(NDF)含量的测定。(I) WT和MsMTA-RNAi株系酸性洗涤纤维(ADF)含量的测定。(J) WT和MsMTA-RNAi株系粗蛋白(CP)含量的测定。
3.3 MsMTA对紫花苜蓿全转录组m⁶A甲基化模式的影响
全基因分布:m⁶A修饰广泛分布于所有染色体且向末端富集,RNAi株系中修饰位点总数发生大幅度减少。
转录本富集:野生型和RNAi株系中m⁶A位点均主要富集于3′UTR区域;基因沉默促使修饰比例进一步向3′UTR偏移。
基序识别:保守序列“GAACU”被鉴定为野生型和RNAi株系中占主导地位的m⁶A识别基序。
基于纳米孔直接RNA测序(ONT-DRS)的全转录组分析揭示,紫花苜蓿的m⁶A修饰广泛分布于染色体末端,并高度富集于转录本的3′非翻译区(3′UTR),且以“GAACU”为核心识别基序。MsMTA的表达抑制不仅导致全基因组水平m⁶A修饰位点总量的急剧下降,还引发了修饰分布从编码区(CDS)向3′UTR的显著偏移,证实了MsMTA在塑造全转录组m⁶A修饰格局中的核心调控地位。(Fig.3)
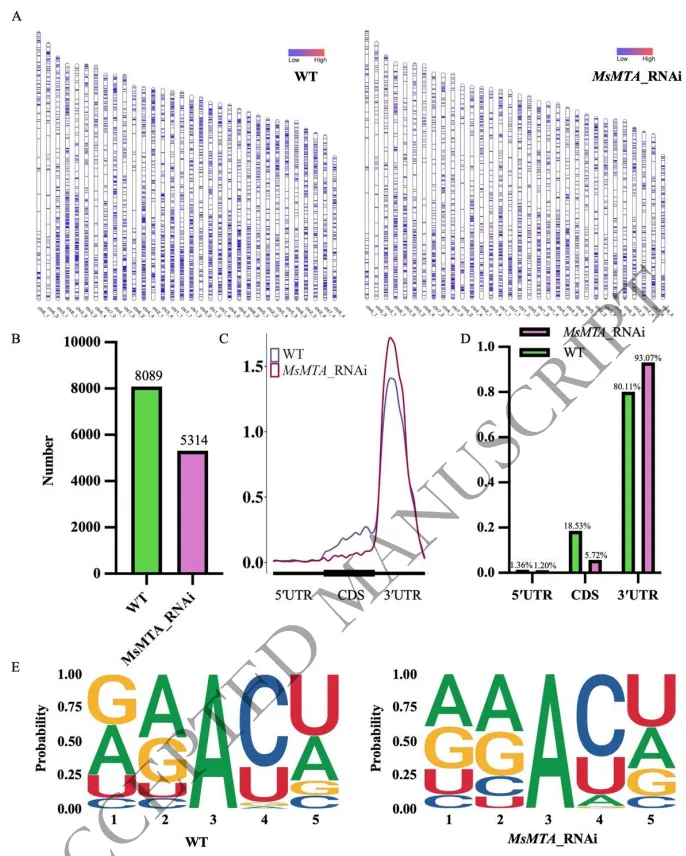
图3. WT和MsMTA-RNAi株系中m⁶A修饰位点的分布分析。(A) 显示WT和MsMTA-RNAi株系中m⁶A修饰位点的分布,颜色梯度代表m⁶A修饰密度,颜色越深表示修饰位点密度越高,颜色越浅表示密度越低。(B) 比较WT和MsMTA-RNAi株系中m⁶A修饰位点数量的条形图。(C) 密度图说明了WT和MsMTA-RNAi株系中5′UTR、CDS和3′UTR区域m⁶A修饰位点的分布,突出了两者之间的差异。(D) 条形图显示了m⁶A峰在不同转录本区域(包括编码区CDS、5′和3′非翻译区UTR以及起始和终止密码子附近)的比例分布,比较了WT和MsMTA-RNAi。(E) 代表WT和MsMTA-RNAi株系样本中主要m⁶A修饰基序的序列标识图。
3.4 MsMTA介导的m⁶A对紫花苜蓿基因表达和代谢通路的调控
转录组改变:RNA-seq鉴定出549个差异表达基因,主要富集于光合作用、细胞壁及木质素生物合成等通路。
修饰组改变:表观转录组分析筛选出434个差异m⁶A修饰基因,绝大多数在RNAi株系中表现为甲基化水平降低。
通路富集:差异修饰基因显著富集于囊泡组织、羧酸代谢、细胞器组装及光合电子传递等关键生物学过程。
转录组与表观转录组的综合分析表明,MsMTA沉默引发了广泛的基因表达重塑和m⁶A甲基化水平下降。差异表达基因高度集中在光合色素代谢与细胞壁结构发育网络中,而差异甲基化基因则主要富集于能量代谢、囊泡运输及有机酸代谢等通路。这些结果表明,MsMTA通过转录与表观转录层面的双重调控,深度参与光合活性与生物量形成所需代谢过程的协调,进而影响植株的茎发育与营养分配。(Fig.4)
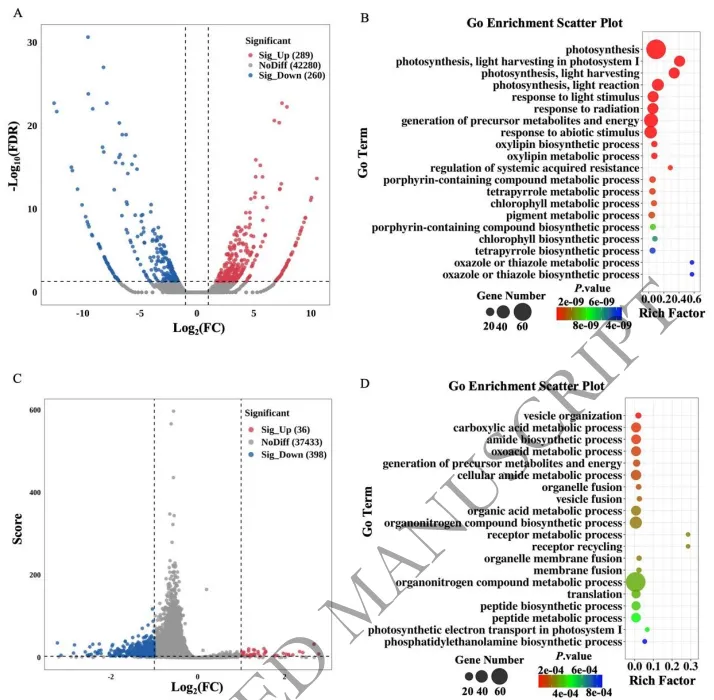
图4. MsMTA-RNAi条件下紫花苜蓿的差异基因表达与m⁶A修饰。(A) 显示紫花苜蓿(WT对比MsMTA-RNAi)差异基因表达的火山图。红点表示显著上调的基因,蓝点表示显著下调的基因。显著性定义为绝对|log₂(Fold Change)| > 1且FDR < 0.05。(B) 差异表达基因(DEGs)的GO富集分析,显示前20个富集的生物学通路。基因比率表示每个通路中DEGs占分配给该通路总基因数的比例。(C) 显示紫花苜蓿(WT对比MsMTA-RNAi)差异m⁶A修饰的火山图。红点代表显著上调的基因,蓝点代表显著下调的基因。显著性定义为绝对|log₂(Fold Change)| > 1且Score > 3。(D) 差异m⁶A修饰基因的GO富集分析,显示前20个富集的生物学通路。基因比率表示每个通路中差异修饰基因占该通路总基因数的比例。
3.5 MsMTA通过m⁶A介导的调控调节氧化还原、小分子代谢和有机氮生物合成通路
联合分析:转录组与甲基化组联合分析发现,多数重叠基因呈现“低甲基化-下调”(Hypo-Down)的表达模式。
代谢重塑:Hypo-Down基因显著富集于氧化还原过程、小分子代谢过程以及有机氮化合物生物合成通路。
核心靶标:包括GS2、SDR1、LQY1等在内的一系列代表性代谢基因表现出m⁶A修饰丢失与转录本丰度下降的协同变化。
将差异表达基因与差异甲基化基因进行交叉比对,发现超过半数的重叠基因表现出甲基化降低伴随表达量下降的“Hypo-Down”模式。这些基因在氧化还原稳态、小分子代谢及氮素同化等关键通路中高度富集。特别是GS2、SDR1、ATPC2等涉及光合电子传递与能量平衡的核心基因,其m⁶A修饰的缺失直接导致了转录本的下调。这表明MsMTA依赖的m⁶A修饰对于维持光合效率及全局细胞代谢稳态具有决定性作用,其功能缺失引发的代谢减弱是导致植株生长受抑的重要驱动因素。(Fig.5)
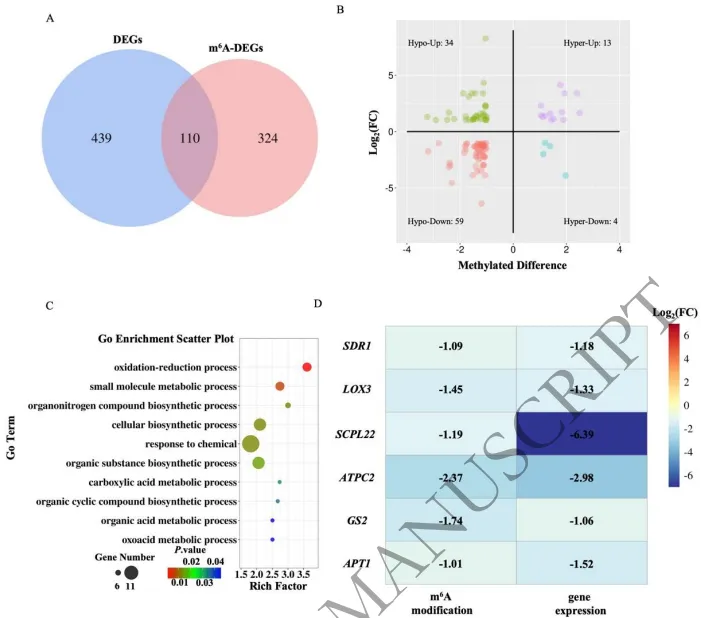
图5. m⁶A修饰与基因表达的关联分析。(A) 差异m⁶A修饰基因(m⁶A-DEGs)与差异表达基因(DEGs)的重叠分析。(B) 说明m⁶A甲基化变化与基因表达之间相关性的象限图。甲基化增加且上调的基因标记为‘Hyper-up’,甲基化增加且下调的基因标记为‘Hyper-down’,甲基化减少且上调的基因标记为‘Hypo-up’,甲基化减少且下调的基因标记为‘Hypo-down’。(C) ‘Hypo-Down’象限中基因的GO富集分析,突出显示富含低甲基化和下调基因的前10个通路。(D) 热图显示参与氧化还原过程、小分子代谢过程和有机氮化合物生物合成过程的差异表达基因(DEGs)的m⁶A甲基化水平与转录本丰度之间的相关性。标注了关键代表性基因,包括短链脱氢酶/还原酶1(SDR1)、脂氧合酶3(LOX3)、丝氨酸羧肽酶样22(SCPL22)、ATP合酶γ亚基2(ATPC2)、谷氨酰胺合成酶2(GS2)和腺嘌呤磷酸核糖转移酶1(APT1)。
3.6 MsMTA-RNAi株系中代表性基因m⁶A富集、转录本水平及mRNA稳定性的验证
富集验证:m⁶A-IP-qPCR证实RNAi株系中MsSDR1、MsGS2等核心代谢基因的m⁶A修饰水平发生显著降低。
表达验证:RT-qPCR结果与测序数据一致,表明上述代表性基因的转录本丰度在RNAi植株中显著下降。
稳定性降:放线菌素D降解试验表明,修饰缺失导致这些关键代谢基因的mRNA降解速率加快、稳定性显著降低。
为了验证高通量测序的可靠性及探究转录后调控机制,采用m⁶A-IP-qPCR和RT-qPCR对三大核心代谢通路中的代表性基因(如MsSDR1、MsLOX3、MsSCPL22、MsATPC2、MsGS2和MsAPT1)进行了定量分析。结果不仅证实了这些基因在MsMTA敲降背景下m⁶A修饰丰度与转录表达量的双重下降,更通过放线菌素D抑制试验明确揭示了其mRNA降解速率的显著加快。综合表明,MsMTA主要通过维持m⁶A依赖的mRNA稳定性来调控下游靶基因的表达,进而掌控紫花苜蓿的全局代谢网络。(Fig.6)
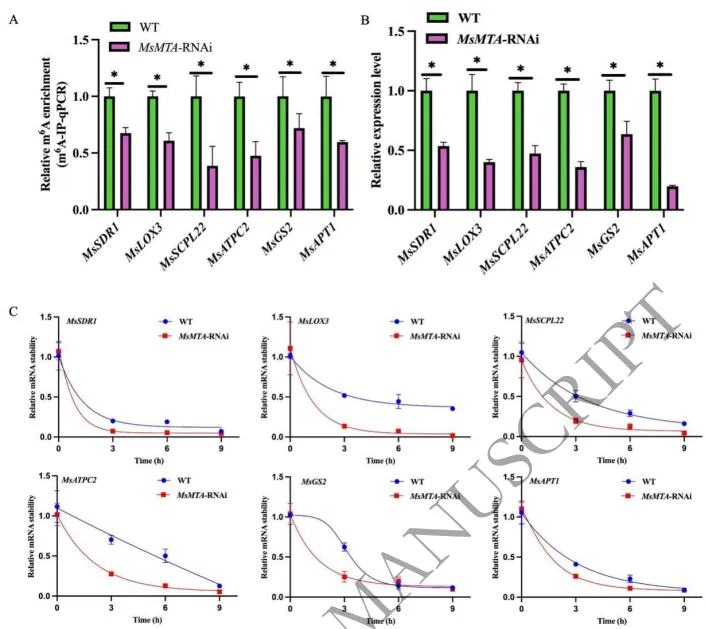
图6. MsMTA-RNAi紫花苜蓿中参与三大主要代谢通路的基因的m⁶A修饰水平、转录本丰度和mRNA稳定性变化的验证。(A) 通过m⁶A免疫沉淀结合定量PCR(m⁶A-IP-qPCR)测定与氧化还原过程(MsSDR1、MsLOX3)、小分子代谢过程(MsSCPL22、MsATPC2)和有机氮化合物生物合成过程(MsGS2、MsAPT1)相关基因的m⁶A富集水平。(B) 使用MsACTIN作为内参基因,通过RT-qPCR定量这些基因的相对表达水平。(C) 为了评估mRNA稳定性,用50 μg/mL放线菌素D处理野生型和MsMTA-RNAi植株,并在不同时间点提取RNA进行RT-qPCR分析。MsACTIN被用作内参。数据以三个生物学重复的平均值±SEM表示。星号表示差异显著(P < 0.05,Student's t检验)。
研究创新性
首次在紫花苜蓿中鉴定并解析m⁶A甲基转移酶功能: 突破了以往紫花苜蓿发育与营养代谢研究多局限于激素或转录调控层面的限制。本研究首次在重要的多年生豆科牧草中鉴定出m⁶A甲基转移酶核心组件MsMTA,并系统证实了其直接控制RNA甲基化模式的核心作用,填补了紫花苜蓿在表观转录组学领域的研究空白。
揭示m⁶A修饰偶联植物生长与碳氮代谢稳态的新机制: 创新性地将微观的RNA表观修饰与宏观的农艺性状(株型、营养品质)直接联系起来。研究发现MsMTA通过特异性维持氧化还原(如MsSDR1、MsLOX3)、小分子代谢(如MsSCPL22、MsATPC2)和有机氮合成(如MsGS2、MsAPT1)等关键代谢基因的mRNA稳定性,来协调光合能量分配与碳氮代谢平衡,提出了m⁶A调控植物生长与代谢网络交汇的全新机制模型。
阐明m⁶A分布偏移决定mRNA稳定性的分子规律: 借助前沿的纳米孔直接RNA测序(ONT-DRS)结合转录组测序,不仅首次绘制了紫花苜蓿全转录组m⁶A图谱,还创新性地发现抑制MsMTA会导致m⁶A修饰位点从编码区(CDS)向3′非翻译区(3′UTR)发生显著偏移。研究证实了这种“低甲基化-下调”(Hypo-Down)模式是加速靶标mRNA降解、削弱整体代谢活性的核心分子基础。
为多年生牧草品质改良提供全新的表观遗传靶点: 传统育种难以兼顾生物量与营养品质的平衡。本研究发现干扰MsMTA表达能够重塑营养物质分配,促使植株形成“高可溶性糖、低纤维、适度低蛋白”的特异性营养特征。这不仅为理解复杂农艺性状的形成提供了新视角,也为未来通过组织特异性或发育阶段特异性地精准操纵m⁶A修饰途径,培育兼顾高产与优质的紫花苜蓿新品种提供了极具潜力的分子靶点和改良策略。

审核|林蘖亙
阅读科研文献,荟萃前沿进展
欢迎关注、转发、投稿、点赞
随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 公开征集35名农业从业者深造一流高校研究生!基层人员均可报名参加!
- 硕泰农业:田间插秧忙 抢抓农时保丰收
- 产教融合新实践 岗位体验育匠心——现代农业系爱宠社团宠物洗护活动
- 招聘信息 || 河北省国富农业投资集团有限公司2026春季招聘
- 山东莒南县:7亿元投资建农业基地,却变身“宴宾楼”“迎宾楼”、星级酒店、健身房,祸国殃民啊!
- Science作者,加入华南农业大学
- 人力资源社会保障部、国家发展改革委、农业农村部联合印发关于统筹城乡就业政策体系的意见
- 产业透视!农业全产业链如何增链、强链、补链?(附中农金旺三大基石全产业链实践)
- 事业单位 | 往届可报: 浙江省绍兴市农业农村局下属事业单位公开招聘高层次人才公告
- 近三年,广东省农业农村厅多名干部被查
