本研究针对传统农药抗药性加剧、多功能生物活性分子匮乏的问题,开发了高效一锅法合成策略,制备了系列新型噻唑并[3,2-a]嘧啶衍生物及嘧啶并[4′,5′:4,5]噻唑并[3,2-a]嘧啶稠环化合物。系统探究了目标化合物的光物理特性、杀虫活性和抗炎活性,结合密度泛函理论(DFT)计算和分子对接技术揭示结构-性能关系,成功筛选出兼具优异杀虫/抗炎活性与聚集诱导发光(AIE)特性的多功能先导化合物,为农药与医药领域的多功能分子研发提供了新范式。
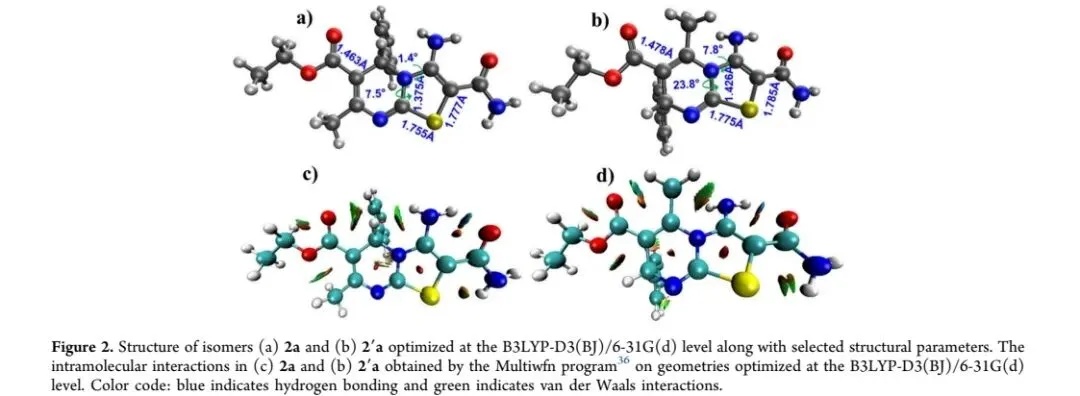
研究以具有广泛生物活性的噻唑并[3,2-a]嘧啶骨架为核心,设计了两步合成路线(Scheme 1):第一步通过Biginelli型三组分反应,以乙酰乙酸乙酯、硫脲和芳香醛为原料,在氯化钙催化下合成4-芳基-3,4-二氢嘧啶-2(1H)-硫酮中间体(1a,b);第二步以中间体1a,b与α-溴氰乙酰胺在碳酸钾存在下经杂环化反应,构建3-氨基噻唑并[3,2-a]嘧啶-2-甲酰胺衍生物(2a,b),并以此为关键前体,通过环化、氯化、胺化、酰化等反应进一步官能化,得到系列稠环化合物(3a,b、4、5a-c、6、7、8)。合成过程中存在两种可能的异构体(2a,b与2′a,b),通过DFT计算证实2a,b为热力学稳定异构体,最终产物经元素分析、FT-IR、¹H NMR、¹³C NMR及EI-MS等技术确证结构。该合成策略具有产率高、反应时间短、操作简便等优势,实现了噻唑并[3,2-a]嘧啶骨架的高效构建与多样化修饰。图一 异构体2a(a)和2′a(b)在B3LYP-D3(BJ)/6-31G(d)水平下的优化结构及部分结构参数;2a(c)和2′a(d)的分子内相互作用(Multiwfn程序计算)。蓝色代表氢键,绿色代表范德华相互作用。图注:该图明确了热力学稳定异构体的结构特征及分子内作用力差异。
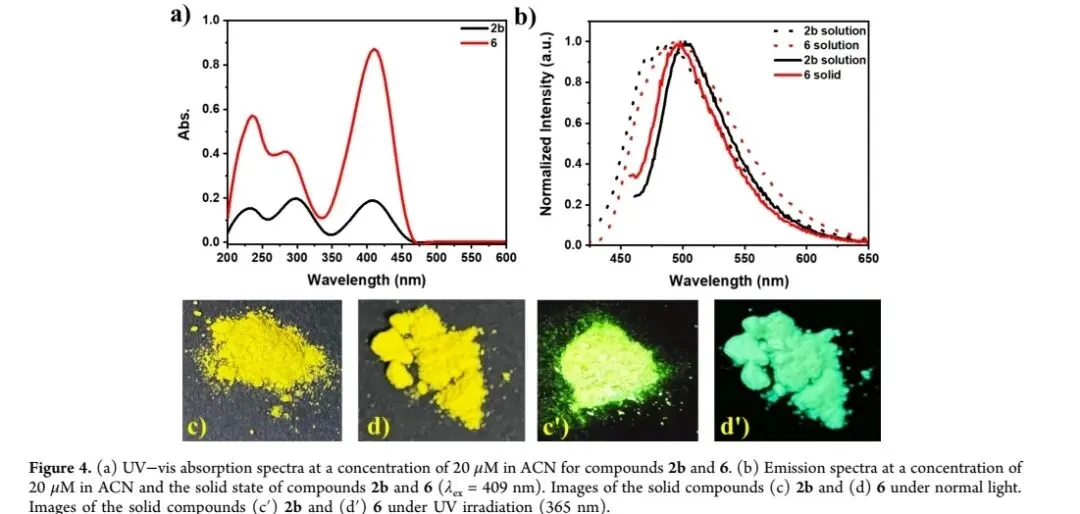
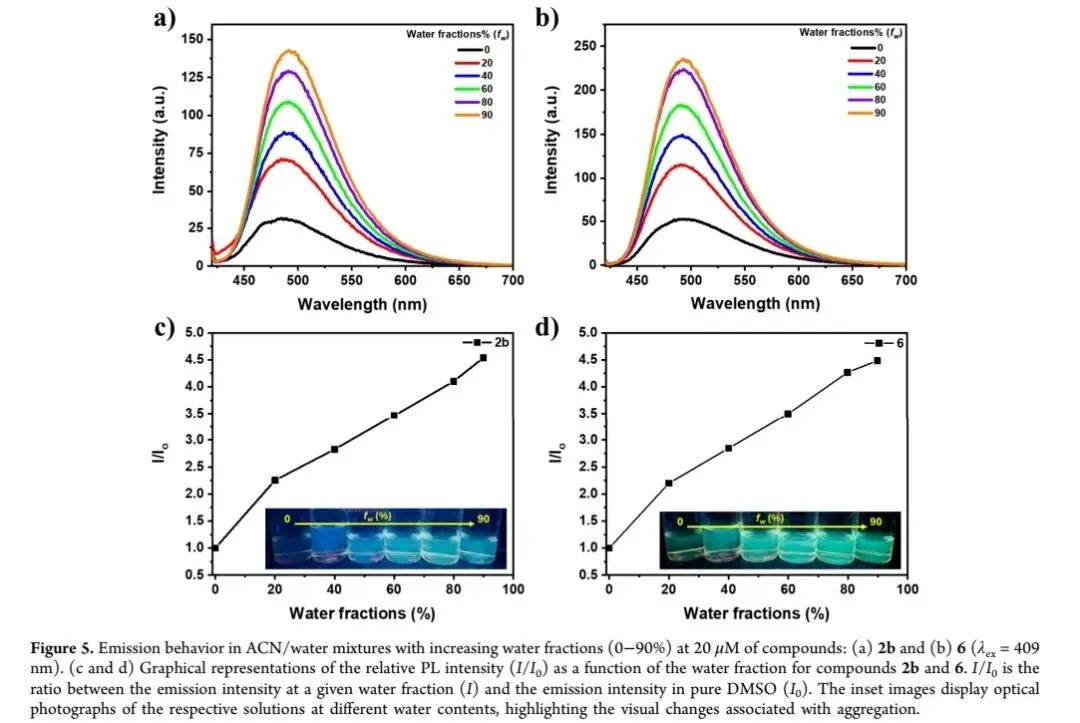
选取化合物2b和6进行光物理特性系统表征,结果显示:在乙腈溶液中(20 μM),两者均表现出特征π-π*跃迁吸收峰,中心波长约409 nm,另在285-298 nm及235 nm处出现辅助吸收峰(图二a)。荧光发射测试表明,化合物2b在溶液中发射峰位于489 nm,固态下红移至502 nm;化合物6在溶液与固态中发射峰均为494 nm,无明显位移,表明其聚集态不发生π-π堆积作用(图二b)。聚集诱导发光(AIE)测试显示,在乙腈/水二元体系中,随着水体积分数从0%增至90%,化合物2b和6的荧光强度逐渐增强,最终达到纯乙腈溶液的5倍(图三a-d),且发射峰位置保持不变,证实其具有典型AIE特性。该现象源于聚集态下分子内运动受限(RIM),减少了非辐射跃迁,提升了辐射衰减效率。此外,化合物2b和6在高水分数条件下的强荧光特性,使其在精准农业荧光示踪领域具有潜在应用价值。图二 化合物2b和6的光物理特性。(a)乙腈溶液(20 μM)中的UV-Vis吸收光谱;(b)乙腈溶液与固态下的荧光发射光谱(λex=409 nm);(c)化合物2b在自然光(左)和365 nm UV光(右)下的固态照片;(d)化合物6在自然光(左)和365 nm UV光(右)下的固态照片。图注:该图展示了目标化合物的吸收/发射特征及固态发光性能。
图三 化合物2b(a、c)和6(b、d)在不同水体积分数的乙腈/水混合体系中的荧光发射光谱(λex=409 nm)及相对荧光强度(I/I0)变化。插图为不同水含量下溶液的光学照片。图注:该图证实了目标化合物的聚集诱导发光特性。
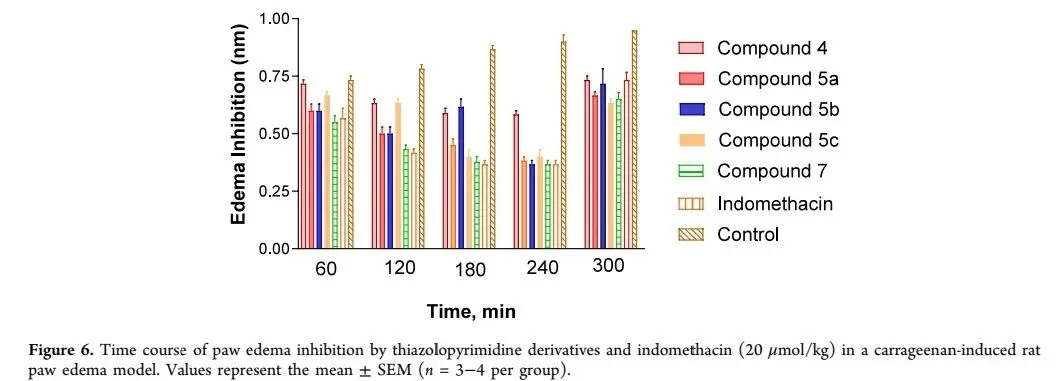
1. 抗炎活性:采用角叉菜胶诱导大鼠足肿胀模型评估化合物抗炎活性,以吲哚美辛为阳性对照。结果显示,所有测试化合物在炎症晚期(2-5 h)均表现出显著抑制作用,表明其可能通过抑制前列腺素合成发挥抗炎作用(图四)。结构-活性关系(SAR)分析表明,C4位取代基是活性关键:氯取代化合物4活性较低(相对吲哚美辛效力46.66%);苯胺取代化合物5a活性提升至68.88%;吗啉取代化合物5c活性最优(71.11%),与吲哚美辛相当;含氯甲基侧链的化合物7活性也达70.00%,为潜在抗炎先导分子。2. 杀虫活性:以棉蚜(Aphis gossypii)若虫和成虫为靶标,评估化合物杀虫活性,以啶虫脒为阳性对照。毒力测定结果显示(表1),化合物5a表现出最强杀虫活性,对若虫的LC₅₀=0.295 mg/L,对成虫的LC₅₀=0.532 mg/L,毒性接近阳性对照(若虫LC₅₀=0.045 mg/L,成虫LC₅₀=0.247 mg/L),毒性比分别为0.152和0.464,显著优于其他衍生物。图四 噻唑并[3,2-a]嘧啶衍生物及吲哚美辛(20 μmol/kg)在角叉菜胶诱导大鼠足肿胀模型中的抗炎时间曲线。数值表示平均值±标准误(n=3-4/组)。图注:该图直观展示了目标化合物的抗炎效果及时间依赖性特征。
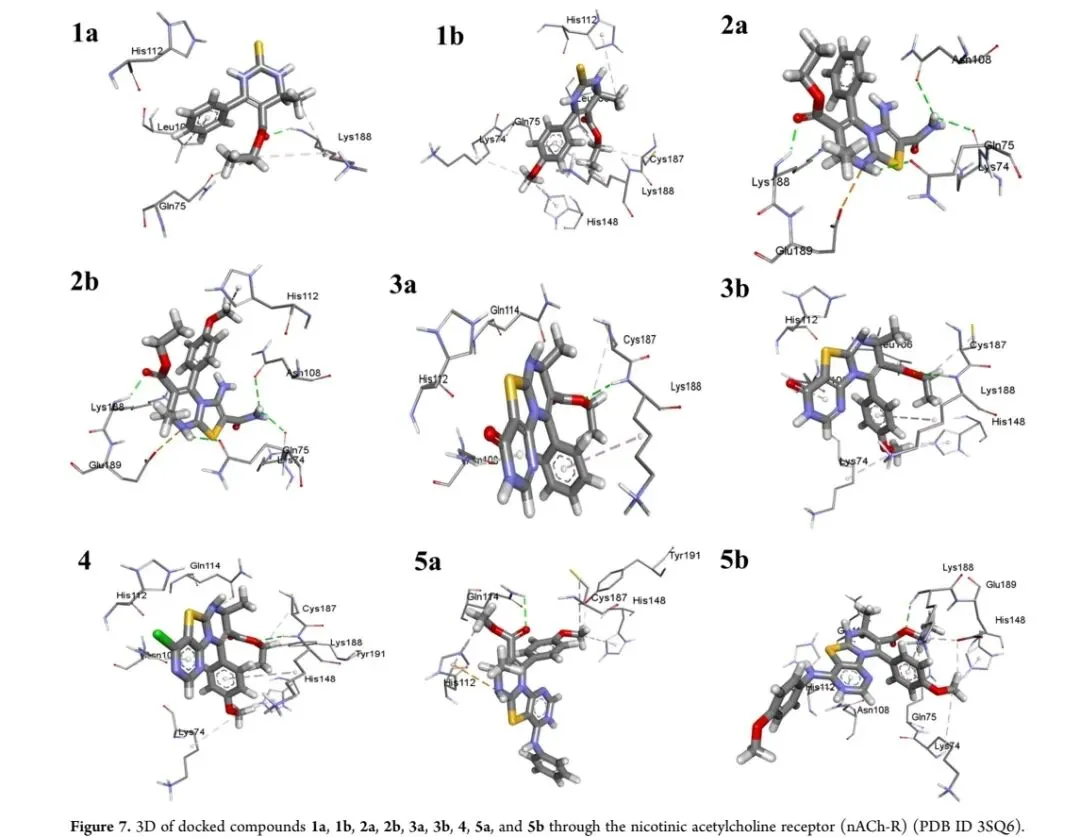
以烟碱型乙酰胆碱受体(nAChR,PDB ID 3SQ6)为靶标进行分子对接,验证杀虫活性机制。结果显示,测试化合物结合能介于-6.00至-7.10 kcal/mol之间,其中化合物8结合能最高(-7.10 kcal/mol),化合物5a与7结合能均为-7.00 kcal/mol(图五、图六)。化合物5a通过与受体关键残基GLN114、TYR191、HIS112等形成稳定氢键和静电相互作用,增强了靶标结合亲和力,与其实验测得的高杀虫活性一致。对接结果表明,C4位苯基氨基取代基可优化分子与受体的疏水和氢键相互作用,是提升杀虫活性的关键结构因素。图五 化合物1a、1b、2a、2b、3a、3b、4、5a、5b与烟碱型乙酰胆碱受体(nACh-R,PDB ID 3SQ6)的3D对接模式。图注:该图展示了配体与受体活性口袋的结合方式。
图六 化合物5c、6、7、8与烟碱型乙酰胆碱受体(nACh-R,PDB ID 3SQ6)的3D对接模式。图注:该图进一步验证了不同取代基对受体结合的影响。
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
