Angew. Chem.| 华中农业大学滕怀龙/武汉大学孔望清团队:光/HAT/镍三重催化实现首例区域、立体和对映选择性C(sp³)-H键单氟烯基化反应
导读:
含氟官能团在药物化学中扮演着至关重要的角色,其中单氟烯烃(monofluoroalkenes)因其独特的性质,既可作为有机合成中的多功能含氟砌块,又常被用作肽键的生物电子等排体。然而,如何高效、高选择性地构建含有手性中心的单氟烯烃,尤其是利用廉价易得的烃类原料,长期是有机合成化学中的一项挑战。近日,华中农业大学滕怀龙教授与武汉大学孔望清教授团队在《Angewandte Chemie International Edition》上合作发表研究论文,首次报道了通过十钨酸四丁基铵盐(TBADT)介导的光诱导氢原子转移(Photo-HAT)与镍催化相结合的协同催化体系,实现了烷基芳烃中苄位C(sp³)-H键与偕二氟烯烃(gem-difluoroalkenes)的区域、立体及对映选择性单氟烯基化反应。该工作使用廉价易得的烃类化合物作为起始原料,在温和的光照条件下,以优异的产率、高达>99/1的E/Z选择性和最高99.5:0.5的对映体比例(er),高效构建了一系列具有叔苄位手性中心的(E)-单氟烯烃类化合物。克级反应及多样的产物衍生化实验展示了该方法的实用性和广阔应用前景。该策略解决了长期以来在C(sp³)-H键直接官能化中同时控制区域、立体和对映选择性的难题,为含氟手性分子的合成开辟了新的路径。
一、研究背景与挑战
1.1 单氟烯烃的重要性
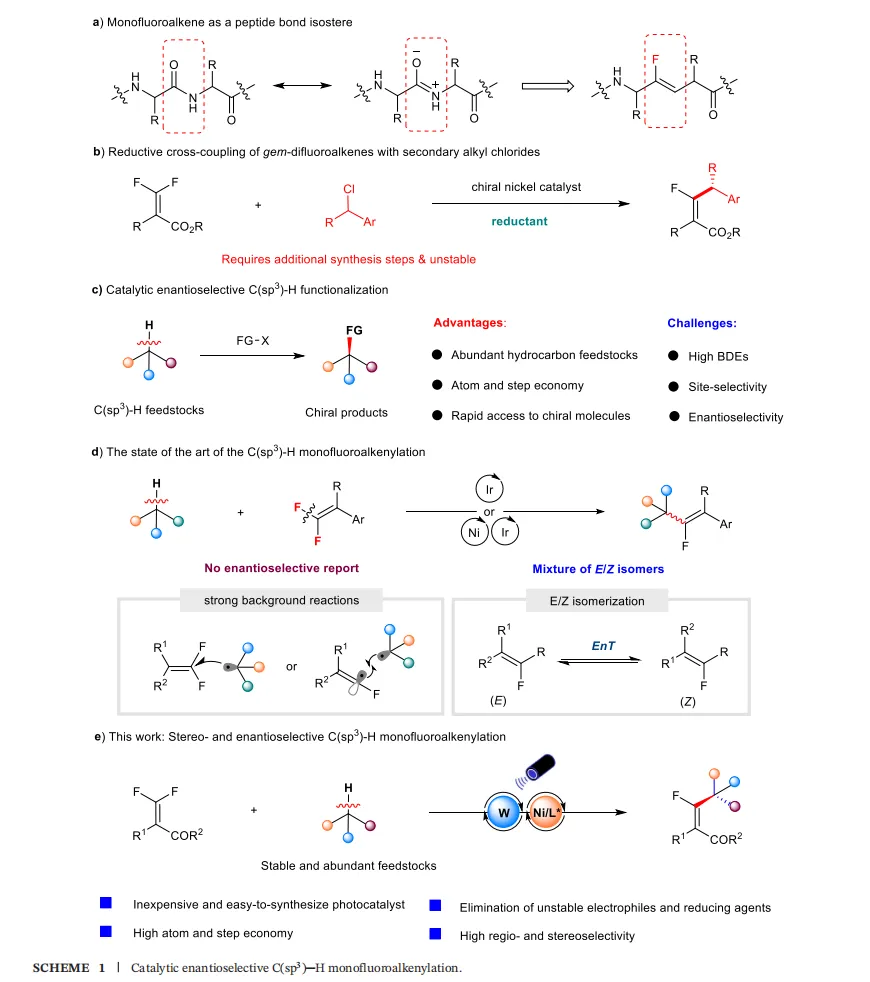
将氟原子或含氟基团引入药物分子中,能显著改善其溶解度、代谢稳定性和生物利用度。单氟烯烃作为一种重要的含氟官能团,因其独特的电子效应和几何构型,不仅是有机合成中常用的含氟砌块,更是药物化学中酰胺键(peptide bond)理想的生物电子等排体,可用于模拟肽键的平面构型和氢键相互作用,在新药研发中具有不可替代的价值(Scheme 1a)。
1.2 现有合成方法的局限
近年来,镍催化的还原偶联反应取得了显著进展,为单氟烯烃的合成提供了有力工具。例如,Fu和Shi等课题组先后报道了利用偕二氟烯烃与烷基亲电试剂(如烷基卤化物)的交叉偶联反应来构建单氟烯烃(Scheme 1b)。然而,这些方法往往依赖于预先制备的、稳定性差且原子经济性不高的烷基亲电试剂。
与之相对,直接对廉价、来源广泛的烃类化合物中的C(sp³)-H键进行官能化,被认为是更为理想和变革性的合成策略(Scheme 1c)。近年来,光诱导氢原子转移(Photo-HAT)与过渡金属催化的结合,已成为实现不对称C(sp³)-H键官能化的强大工具,并成功应用于C(sp³)-H键的芳基化、烯基化、酰基化等反应中。
1.3 本工作面临的挑战
尽管前景广阔,但实现C(sp³)-H键的直接单氟烯基化反应面临着多重严峻挑战(Scheme 1d):
反应路径复杂:从底物烃生成的烷基自由基,既可以与偕二氟烯烃发生自由基加成,也可能引发单电子还原形成自由基负离子,进而发生C-F键断裂。多种反应路径并存,导致选择性控制极难。
背景反应竞争:这些自由基介导的背景反应通常缺乏选择性,容易产生难以分离的混合物。
E/Z异构化问题:光化学反应中普遍存在的能量转移过程,容易导致C=C双键的E/Z异构化,使得产物立体选择性(几何构型)的控制难上加难。
(图源: Angew. Chem. Inr. Ed.)
二、本工作策略:Photo-HAT与镍催化的协同
面对上述挑战,华中农业大学滕怀龙教授与武汉大学孔望清教授团队设想:能否利用氢原子转移(HAT)光催化剂(如TBADT)选择性地断裂烃类底物中电子丰富或空间位阻较小的C(sp³)-H键,生成相应的烷基自由基;然后,利用手性镍催化剂高效、高选择性地捕获该自由基,并促使它与偕二氟烯烃发生偶联,最终通过β-F消除步骤,实现对映选择性和立体选择性的单氟烯基化(Scheme 1e)。
该策略的关键优势在于:
三、反应条件优化
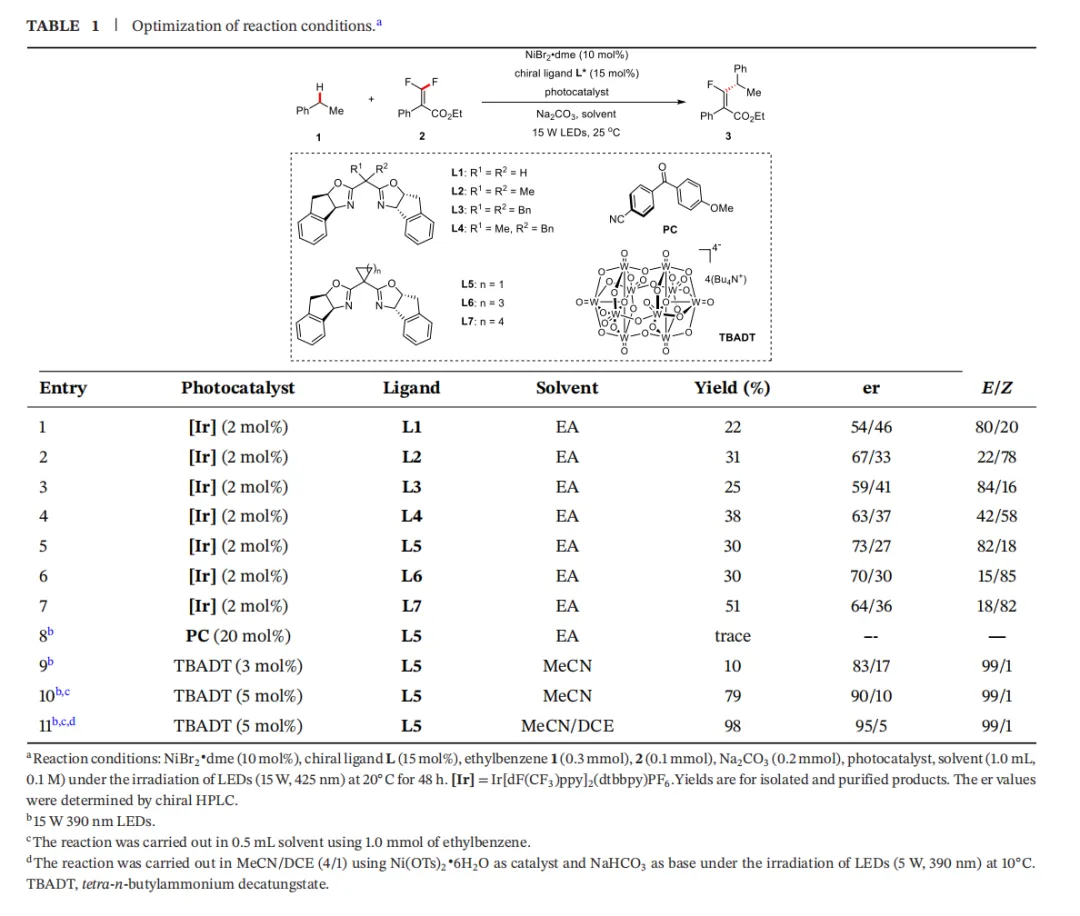
研究者以乙苯(1)和偕二氟烯烃(2)为模型底物,对反应条件进行了系统的筛选。
关键发现与优化历程:
初步尝试与配体筛选:使用Ir光敏剂和NiBr₂/dme为镍源,初步能得到目标产物,但产率和选择性均不理想。随后对一系列手性配体(L1-L7)进行了筛选,结果显示,环丙烷骨架的手性配体L5在反应性和对映选择性方面表现最优。
光催化剂的重大突破:当用廉价、易得的TBADT替代昂贵的Ir光敏剂时,反应的立体选择性(E/Z)得到了质的飞跃,从之前的约80/20提升至>99/1。
反应参数的精细调控:进一步优化发现,溶剂的极性、反应浓度、镍源(Ni(OTs)₂·6H₂O)和碱(NaHCO₃)对产率和对映选择性有显著影响。最终确定的最优条件为:Ni(OTs)₂·6H₂O (10 mol%), L5 (15 mol%), TBADT (5 mol%), NaHCO₃ (2 equiv), MeCN/DCE (4/1) 混合溶剂, 390 nm LED光照, 10°C反应48小时,能够以98%的分离产率,95:5的er值和>99/1的E/Z比得到目标产物。
(图源: Angew. Chem. Inr. Ed.)
四、底物范围拓展
该方法展现出卓越的底物普适性和官能团兼容性。
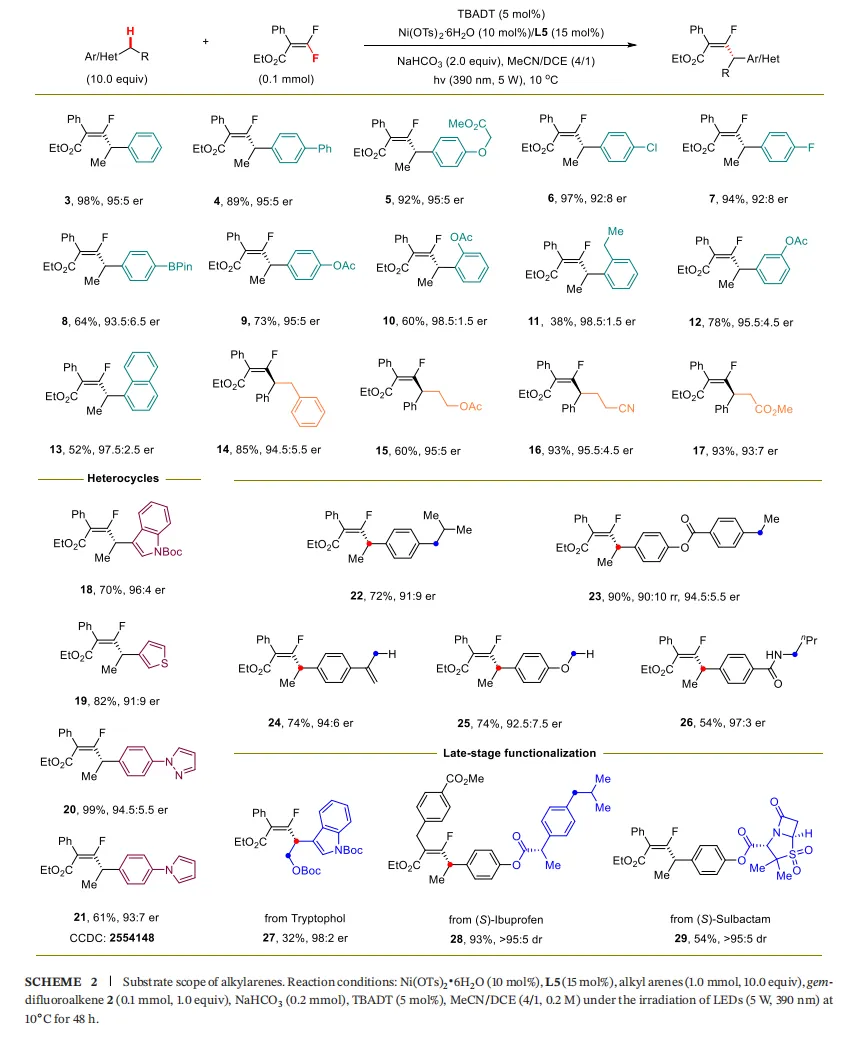
4.1 烷基芳烃底物(Scheme 2)
电子效应与位阻效应:苯环上带有不同电子性质(给电子、吸电子)和空间位阻的取代基的乙苯衍生物,均能顺利反应,以良好的产率(42-85%)和优异的对映选择性(高达97:3 er) 得到目标产物(3-12)。
杂环化合物:一系列药物相关的杂环如吲哚、噻吩、吡唑、吡咯等,均能很好地兼容(18-21)。
化学选择性:
复杂分子后期修饰:该方法成功应用于天然产物和药物分子的后期修饰,如色醇(27)、(S)-布洛芬(28)和(S)-舒巴坦(29)的衍生化,以良好的产率和优异的立体选择性得到目标产物,展示了其在药物化学中的巨大潜力。
(图源: Angew. Chem. Inr. Ed.)
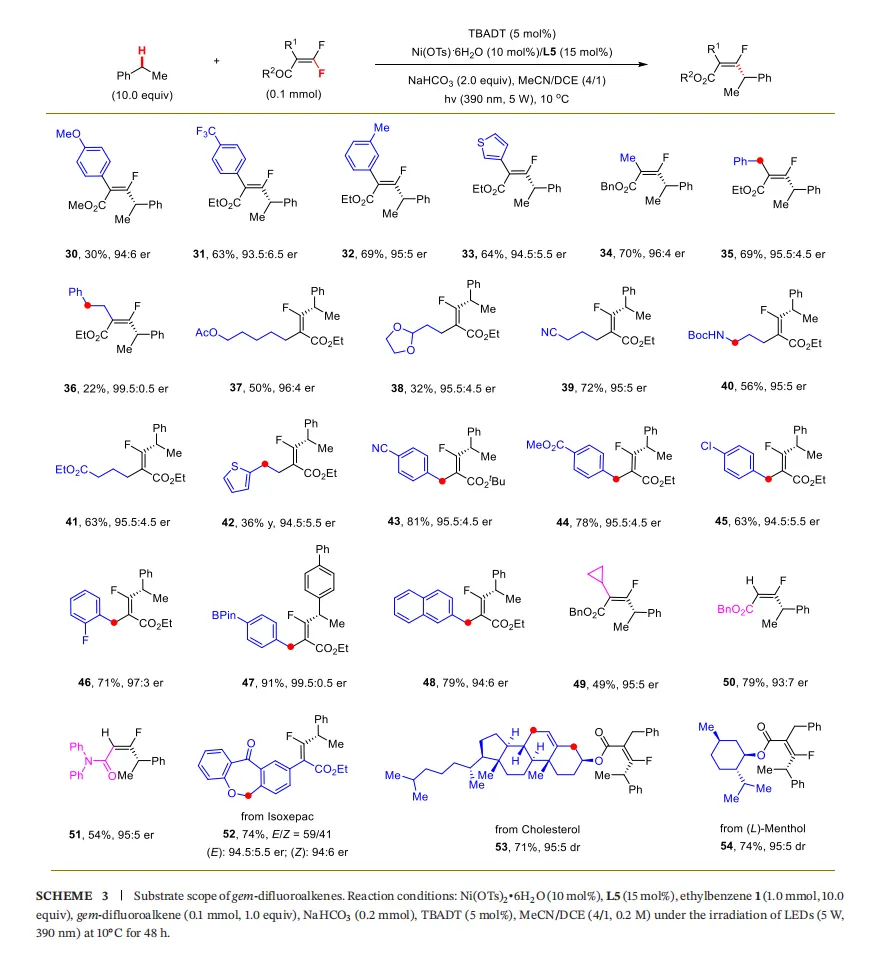
4.2 偕二氟烯烃底物(Scheme 3)
芳基取代:苯环上带有不同电性及位阻基团的偕二氟烯烃均可高效转化(30-32),杂芳基(噻吩)取代底物同样适用(33)。
烷基取代:烷基链上带有芳基、酯基、缩醛、氰基、酰胺、噻吩等多种官能团的底物均能很好地耐受,以中等到优良的产率和高达96:4的er值得到产物(35-42)。
苄基取代:不同电性和位阻的苄基取代底物也能顺利反应(43-48),且氯和硼酸酯基团可以被兼容,为后续衍生化提供了方便(45, 47)。
挑战性底物:环丙烷取代的底物(49)及含酯基、酰胺基的三取代偕二氟烯烃(50, 51)同样适用,未检测到环丙烷开环副产物。
复杂分子衍生:从异克塞平(isoxepac)、胆固醇和薄荷醇衍生的偕二氟烯烃,均能以良好的产率和优异的非对映选择性得到目标产物(52-54),进一步证明了该方法的普适性和实用性。
(图源: Angew. Chem. Inr. Ed.)
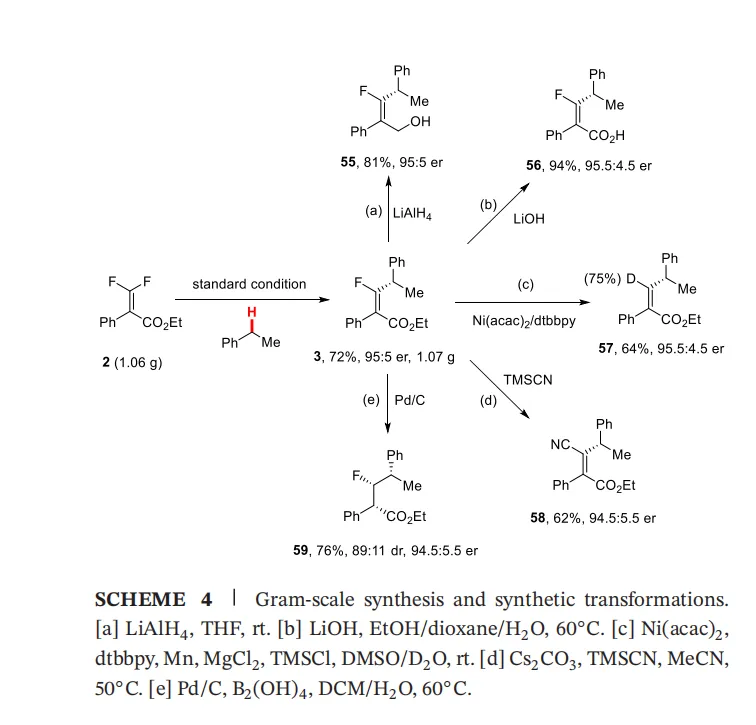
五、克级反应与合成应用(Scheme 4)
(图源: Angew. Chem. Inr. Ed.)
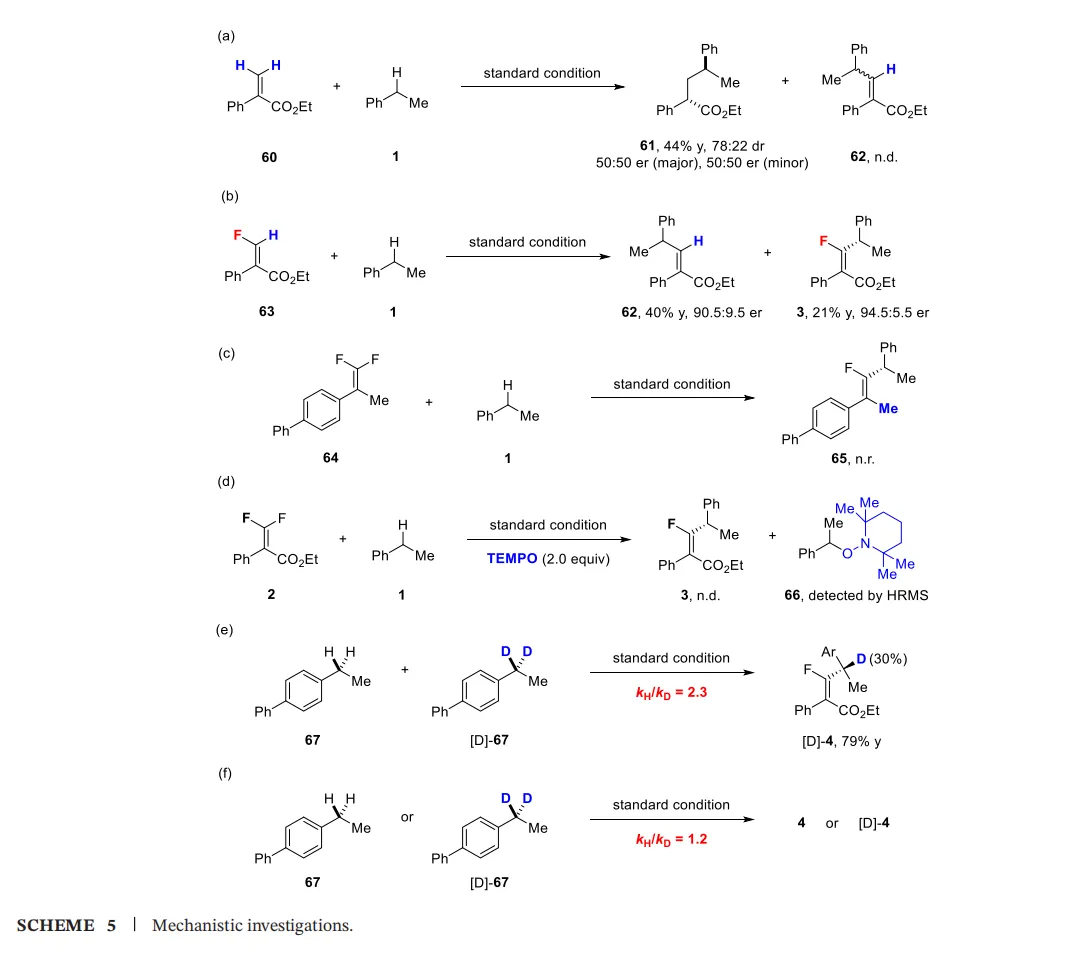
六、机理研究(Scheme 5)
为深入探究反应机理,研究者进行了一系列控制实验(Scheme 5)。
氟原子的关键作用:当使用非氟代的丙烯酸酯底物(60)时,仅得到外消旋的Giese加成产物(61)。这表明底物中的氟原子对实现高对映选择性的交叉偶联至关重要,可能通过与金属中心的独特配位方式影响了反应路径。
E-单氟烯烃的反应性差异:合成并分离的(E)-单氟烯烃63在标准条件下反应,除得到目标产物62外,还得到了脱氢烷基化产物3。这与偕二氟烯烃只生成(E)-产物的结果截然不同,暗示单氟烯烃可能经历烷基-镍中间体的迁移插入及随后的β-F消除机理,而非简单的自由基偶联。
排除SET还原机理:前人报道中,偕二氟烯烃可被光催化还原生成单氟烯基自由基。但在本体系中,当使用易被还原的底物64时,并未得到预期的偶联产物65,有力地排除了该反应经由单氟烯基自由基中间体的可能性。
自由基捕获实验:加入TEMPO后,反应被完全抑制,并通过HRMS检测到苄基自由基被捕获的产物66,直接证实了反应体系中苄基自由基的生成。
动力学同位素效应(KIE):
可能的催化循环
基于机理实验和前人的研究,作者提出了如下可能的催化循环:
HAT过程:光激发态*[W₁₀O₃₂]⁴⁻(TBADT)选择性地攫取烷基芳烃(1)中苄位C-H键的氢原子,生成苄基自由基(I)和还原态的[HW₁₀O₃₂]⁴⁻。
Ni(I)生成:Ni(II)前驱体被还原态的TBADT或体系中的碱(如HCO₃⁻)还原为Ni(0)物种,Ni(0)进一步与手性配体L5配位。
自由基捕获:苄基自由基(I)被手性Ni(I)络合物捕获,形成Ni(II)-烷基络合物(II)。
C-F键活化与偶联:Ni(II)-烷基络合物(II)与偕二氟烯烃(2)发生配位/迁移插入,生成Ni(II)中间体(III)。
β-F消除:中间体(III)发生区域选择性和立体专一性的顺式β-F消除,释放出目标(E)-单氟烯烃产物(3),同时再生Ni(0)催化剂,完成催化循环。
(图源: Angew. Chem. Inr. Ed.)
七、总结与展望
华中农业大学滕怀龙与武汉大学孔望清团队合作,报道了首例通过TBADT介导的Photo-HAT与手性镍催化协同作用,实现烷基芳烃苄位C(sp³)-H键与偕二氟烯烃的区域、立体及对映选择性单氟烯基化反应。
核心创新点:
策略突破:首次实现了C(sp³)-H键的直接、高选择性单氟烯基化,解决了该领域长期存在的反应性和选择性难题。
精准调控:通过廉价的TBADT光催化剂与手性镍催化剂的协同,实现了对HAT过程、自由基捕获和C-F键断裂的精准控制,从而同时掌控了反应的区域、立体(E/Z)和对映选择性。
绿色经济:使用易得的烃类为原料,避免了传统方法中不稳定亲电试剂和过量还原剂的使用,具有极高的原子经济性和步骤经济性。
底物范围广:适用于多种烷基芳烃和偕二氟烯烃,包括复杂药物分子和天然产物的后期修饰,展现了优异的官能团耐受性和化学选择性。
实用性强:克级反应和多样的产物衍生化证明了该方法在合成含氟手性药物和生物活性分子中的重要应用潜力。
未来发展方向:
挑战与展望:尽管对苄位C-H键取得了成功,但对于普通烷烃中更具挑战性的非活化C(sp³)-H键的不对称官能化仍需进一步探索。
机理研究:更深入的DFT理论计算和详细的动力学研究,有助于更清晰地揭示手性诱导和β-F消除的具体机制,为配体设计提供理论指导。
新反应拓展:该策略有望拓展至其他类型的偶联 partners,例如构建含有C-F键的季碳手性中心,进一步丰富含氟手性分子的合成工具箱。
该工作为热力学稳定且反应活性低的C(sp³)-H键的直接不对称官能化提供了一个强有力的新策略,对药物化学、农药化学及含氟功能材料领域具有重要的推动作用。
论文信息
题目: Regio-, Stereo-, and Enantioselective C(sp³)-H Monofluoroalkenylation via Photo-HAT/Nickel Dual Catalysis
作者: Yate Chen, Chengbing Yang, Zhaojing Xu, Zidong Liang, Yifan Xu, Wangqing Kong, Huailong Teng
期刊: Angewandte Chemie International Edition, 2026
DOI: 10.1002/anie.2026201
说明: 以上内容基于已发表学术论文进行整理与解读,版权归原作者与出版社所有。仅供学术交流与参考。