丨旨在分享学术交流丨能力有限欢迎指正丨
「上海岱算科技有限公司」已向境内外230余家高等院校/科研院所提供了累计1400多项模拟计算服务,赋能科学研究提速增效!合作实验课题组在线发表学术论文期刊有ACS系列、AM系列、Angew、CEJ、EST、JACS、Matter、Nature子刊等,助力科研工作锦上添花!理解并调控锂硫电池(LSBs)的速率决定步骤(RDSs)对于提升其电化学性能至关重要。
2026年05月18日,河南农业大学吴俊锋、郑州大学/南开大学周震、南开大学/郑州大学唐宾团队合作在Advanced Materials期刊发表题为“Solid-Liquid Synergy Enables a Trisulfur-Radical-Rich Microenvironment for Accelerated Li-S Conversion Kinetics”的研究论文,河南农业大学Zhao Zhiqi、河南农业大学/南开大学Zhang Bohai为论文共同第一作者,吴俊锋、周震、唐宾为论文共同通讯作者。
第一作者:Zhao Zhiqi、Zhang Bohai
通讯作者:吴俊锋、周震、唐宾
论文DOI:10.1002/adma.73407
该研究提出了一种协同策略,将含有氧空位的模板硫宿主与作为传统醚基电解液添加剂的高供体数(高DN)溶剂相结合。该策略在正极侧构建了一个局域高DN微环境,其中含有大量三硫自由基。实验和计算均证实,三硫自由基作为关键介质,可将RDS从本质上缓慢的准液-固反应加速至动力学上更有利的三硫自由基介导的转化过程。得益于三硫自由基介导的RDS增强,该LSB在1 C倍率下循环500圈后仍保持85.4%的容量,平均每圈衰减率仅为0.03%。此外,在5 C倍率下实现了659.6 mAh g⁻¹的初始容量,或在4.6 mg cm⁻²的高硫载量下实现了1126.9 mAh g⁻¹的比容量。该研究提出了一种新型的三硫自由基介导的催化机制,并通过界面工程与电解液调控相结合,突破了固有RDS限制。
锂硫电池(LSBs)因其高能量密度(>700 Wh kg⁻¹)和硫正极高达1672 mAh g⁻¹的理论比容量,被视为有前景的下一代储能系统。LSBs的硫氧化还原反应(SRR)涉及从S₈到溶解的多硫化物中间体Li₂Sₓ(2 < x ≤ 8),并最终转化为Li₂S的多步固-液-固转化过程。在这些步骤中,从Li₂S₄到绝缘Li₂S的准液-固转化被广泛认为是SRR中的速率决定步骤(RDS)。该RDS不仅贡献了三分之二的理论容量,还决定了电极界面的稳定性和倍率性能。此外,RDS的缓慢动力学减慢了长链多硫化物(如Li₂S₆)的前期电化学转化,导致其累积并引发严重的穿梭效应。因此,优化RDS的动力学对于实现高效SRR和LSBs的循环稳定性至关重要。
LSB催化剂有望加速准液-固转化过程,是解决上述挑战最高效的方案之一。在近期研究中,吸附-催化材料如金属化合物、单原子催化剂、异质结、缺陷工程结构和掺杂工程结构因其对Li₂S₄物种的强极性而被广泛研究,能够实现对其锚定并促进其转化为不溶性产物。然而,尽管传统的醚基电解液(即1,2-二甲氧基乙烷(DME)和1,3-二氧戊环(DOL)按1:1体积比混合,并加入1 m双三氟甲烷磺酰亚胺锂(LiTFSI)和2%硝酸锂(LiNO₃))因与锂负极良好的相容性和适中的粘度而在LSBs中广泛使用,但它们仅能中等程度地溶剂化长链多硫化物,限制了液-液转化的效率。此外,短链多硫化物由于比长链多硫化物的溶解度低得多,容易沉淀并在正极表面形成绝缘Li₂S₂/Li₂S沉积物。这些沉积物通常发展为覆盖电极表面的(二维)钝化膜。因此,迫切需要合理设计先进的催化体系,例如加速RDS、调控Li₂S沉积以及增强多硫化物溶解度,以突破这些限制并加速整体反应动力学。近期研究表明,通过调控催化剂表面受控的溶剂吸附来设计双电层(EDL)是维持稳固EDL结构的有效策略,从而促进多硫化物传输和电子转移。
三硫自由基(S₃∙⁻)作为一种高活性中间体,能显著加速硫物种的相互转化,有望从根本上促进LSBs中SRR的RDS动力学。高供体数(DN)溶剂,包括1,3-二甲基-2-咪唑烷酮(DMI)、四甲基脲(TMU)、二甲基亚砜(DMSO),可以稳定该自由基,同时调控Li₂S的沉积并显著增强多硫化物的溶解度。然而,高DN溶剂通常与锂负极的相容性差且粘度高。为解决这些问题,通常将高DN溶剂的含量控制在最低,并用作传统醚基电解液的添加剂。某些高DN溶剂分子可以被醚基溶剂分子包围,形成核-壳溶剂化结构,提供空间屏蔽以保护锂负极,同时仍能稳定S₃∙⁻。这些发现极大地促进了对通过本体电解液中的高DN溶剂稳定S₃∙⁻及其对锂硫电池性能有益影响的理解。除了溶剂化化学,另一种稳定活性硫物种的物理途径已出现,该途径受洪德规则支配。这种方法能够在铁磁性衬底上形成超稳定的三重硫自由基对([Sₓ∙⁻−−Sₓ∙⁻]),为获得自由基介导的反应动力学提供了一条新途径。
在此,该研究提出了一种新型隔离策略,通过构建由羰基(C=O)孤对电子与氧空位(OVs)相关的金属位点之间配位介导的正极局域高供体数(高DN)溶剂微环境,整合了化学和物理效应。作为含C=O的代表性高DN溶剂,将DMI(0.7 vol%)引入传统醚基电解液以稳定S₃∙⁻。采用具有丰富OVs的Co掺杂Fe₂O₃作为模板硫宿主。该设计最初用于增强多硫化物溶解度,同时减轻穿梭效应。出乎意料的是,原位电化学测量和理论计算共同揭示了S₃∙⁻在加速速率决定步骤(RDS)中的催化作用。因此,该LSBs表现出良好的长循环和倍率性能。在1 C倍率下循环500次后容量保持率为85.4%,平均每圈衰减率仅为0.03%。此外,在5 C倍率下初始容量高达659.6 mAh g⁻¹。基于热力学和动力学验证的S₃∙⁻介导的催化机制有望为提高LSBs的能量密度提供一条有前景的途径。
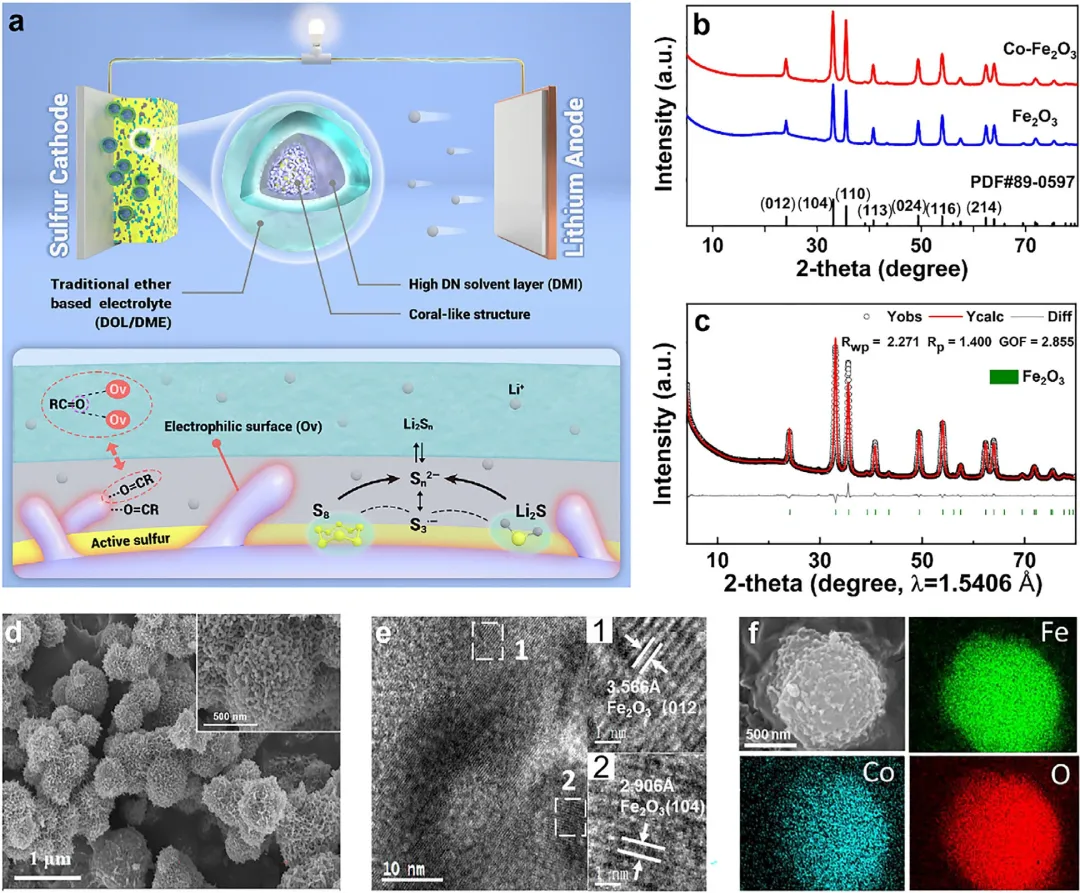
图1 机理与表征。(a) 所提出策略及催化机理示意图;(b) 合成的Fe₂O₃和Co-Fe₂O₃催化剂的XRD图谱;(c) Co-Fe₂O₃催化剂的XRD图谱Rietveld精修;(d) Co-Fe₂O₃催化剂的SEM图像;(e) Co-Fe₂O₃催化剂的HRTEM图像;(f) Co-Fe₂O₃催化剂的元素分布图。
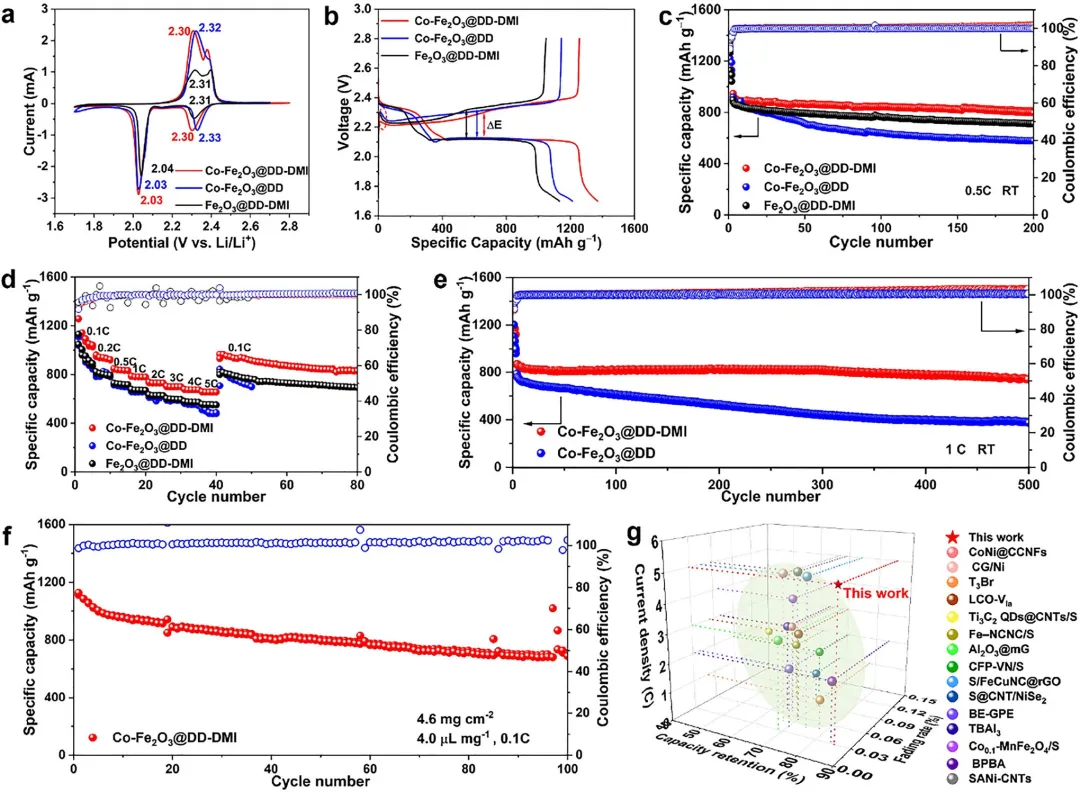
图2 电化学性能测试。(a) 扫描速度为0.1 mV s⁻¹时的CV曲线;(b) 0.1 C倍率下首圈的充放电曲线;(c) 0.5 C倍率下的循环性能;(d) 倍率性能;(e) 1.0 C倍率下的长循环性能;(f) Co-Fe₂O₃@DD-DMI在0.1 C倍率下、高硫载量4.6 mg cm⁻²时的循环性能;(g) 该研究与已报道研究的性能对比。
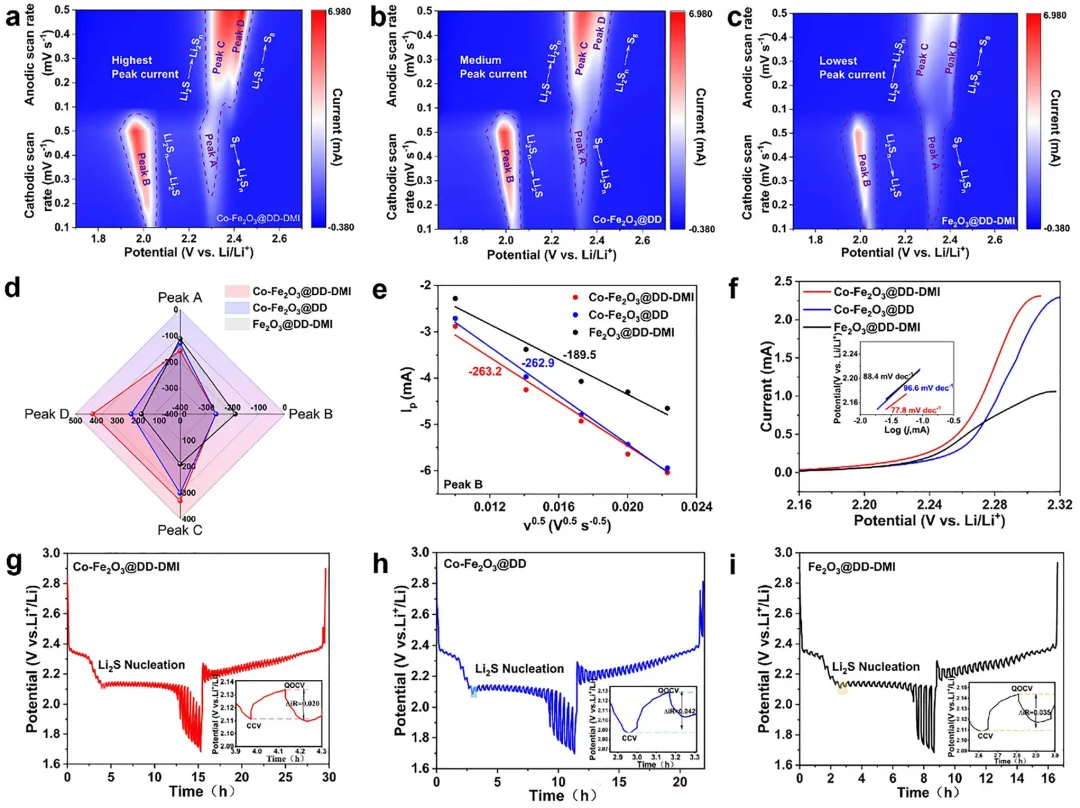
图3 Co-Fe₂O₃@DD-DMI、Co-Fe₂O₃@DD和Fe₂O₃@DD-DMI电池的电化学行为测试。(a-c) 不同扫描速率下的CV曲线;(d) CV曲线四个氧化还原峰拟合斜率系数的雷达图;(e) B峰的拟合;(f) Tafel斜率;(g-i) GITT曲线。
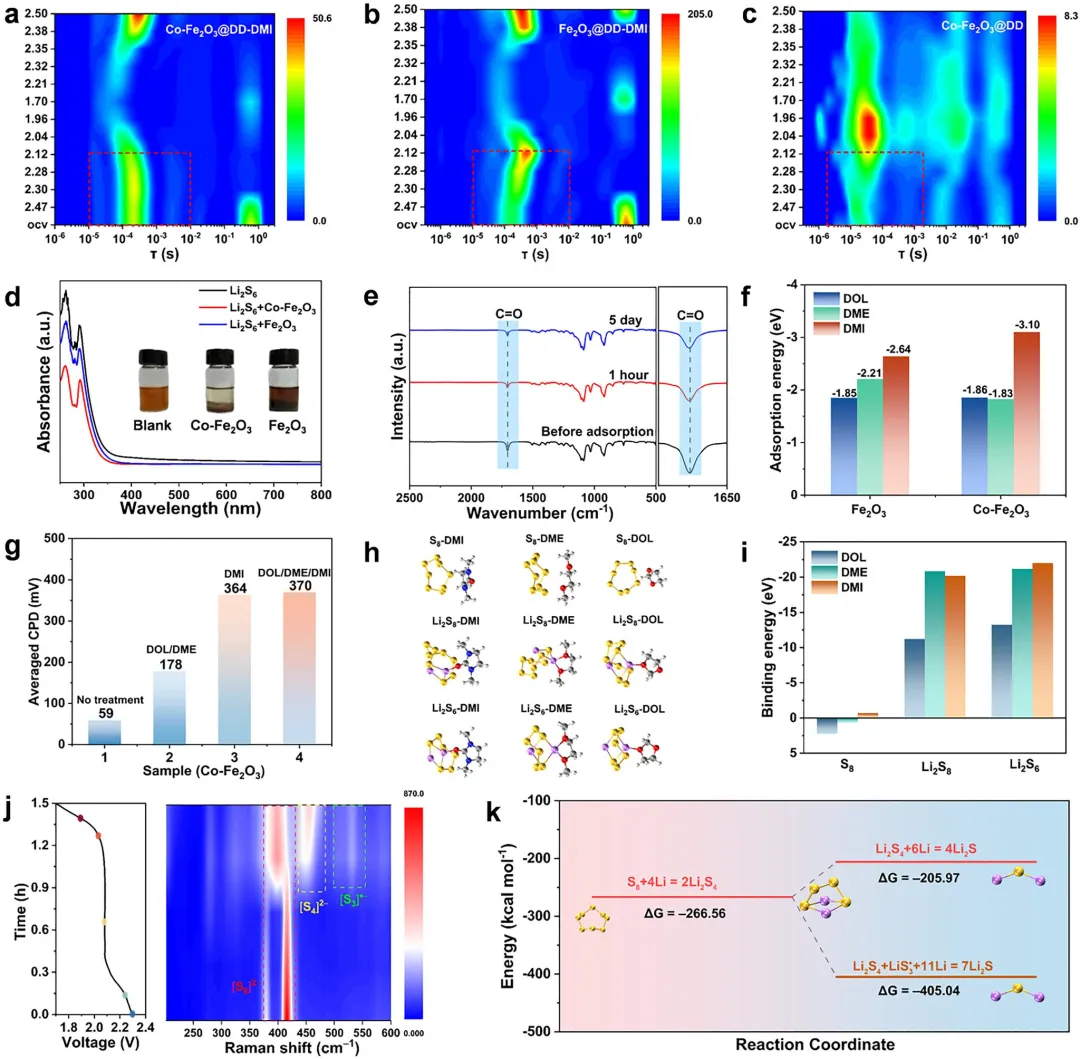
图4 (a-c) 放电过程中Co-Fe₂O₃@DD-DMI、Co-Fe₂O₃@DD和Fe₂O₃@DD-DMI电池的DRT谱图演化;(d) 不同样品对Li₂S₆吸附后的UV-vis光谱及光学照片(插图);(e) 与Co-Fe₂O₃相互作用前后DMI的FTIR谱图;(f) 不同溶剂分子在Fe₂O₃和Co-Fe₂O₃表面的吸附能计算值;(g) 不同样品表面的平均接触电位差(CPD)对比;(h, i) Li₂S₈和S₈与DMI分子的结合能计算及优化构型;(j) Co-Fe₂O₃@DD-DMI电池放电过程中的原位拉曼光谱;(k) Li₂S₄转化为Li₂S的传统路径与S₃∙⁻介导路径的吉布斯自由能变化对比。
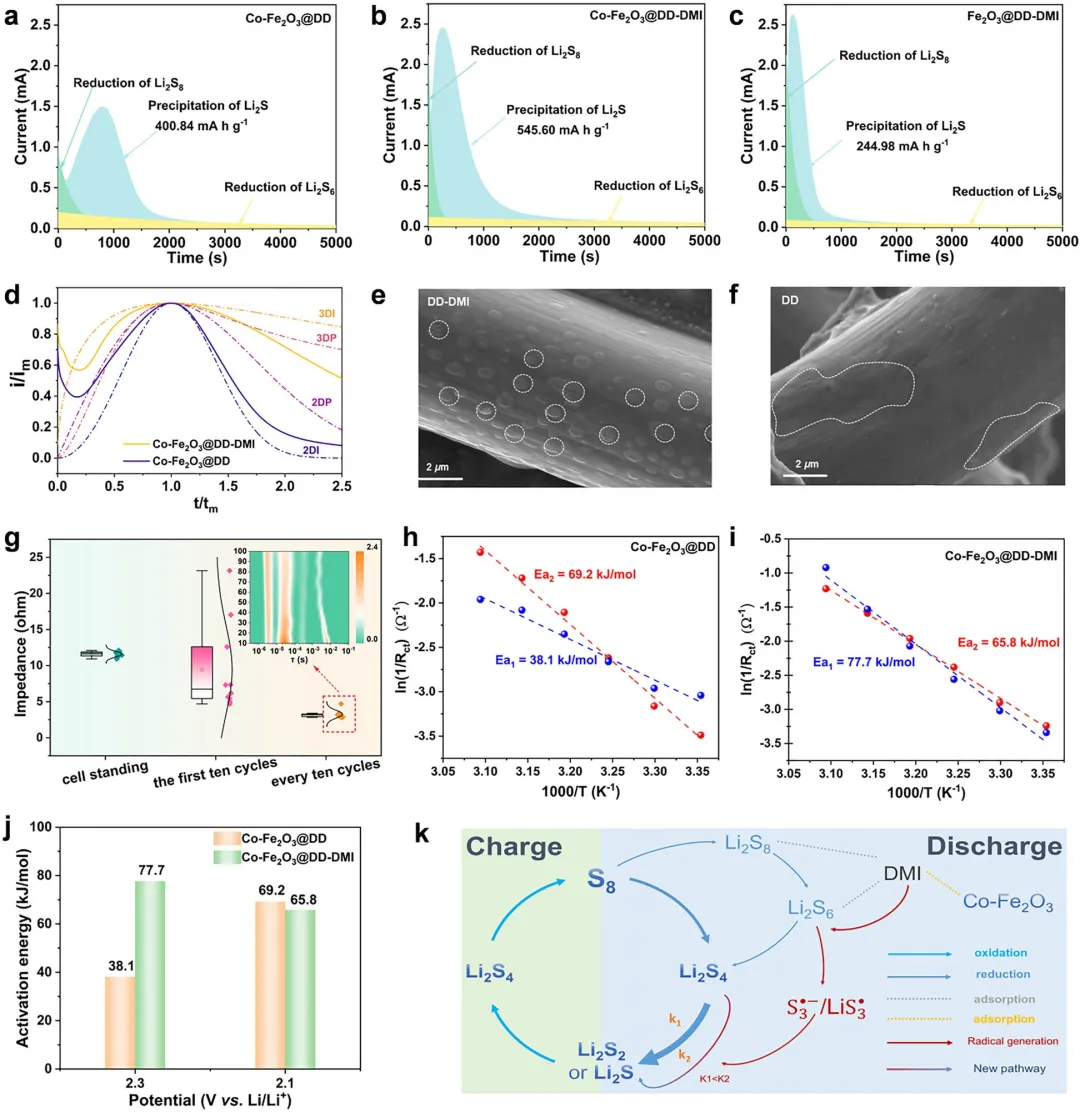
图5 (a) Co-Fe₂O₃@DD,(b) Co-Fe₂O₃@DD-DMI,和(c) Fe₂O₃@DD-DMI电池中 Li₂S成核动力学;(d) Li₂S的成核模型以及(e, f) Co-Fe₂O₃@DD-DMI和Co-Fe₂O₃@DD电池中Li₂S电沉积的相应SEM图像;(g) 随着电池静置、前10个循环以及随后每10个循环的电化学阻抗演化,插图为DRT分析;(h) Co-Fe₂O₃@DD电池和(i) Co-Fe₂O₃@DD-DMI电池在第一和第二放电平台通过Arrhenius方程(Rct来自变温EIS测量)推导表观活化能;(j) 从图5h,i中提取的活化能比较;(k) 催化剂与电解液协同作用下的转化路径示意图,突出了DMI吸附、S₃∙⁻形成以及SRR动力学的调控。
总之,该研究展示了一种调控SRR中RDS的新型且有效的方法。通过构建局域高DN溶剂微环境以稳定高活性S₃∙⁻,有效加速了传统上缓慢的Li₂S₄ → Li₂S转化,从而缓解了硫还原固有的动力学瓶颈。因此,该体系表现出更快的反应动力学,并增强了理想的三维Li₂S沉积可逆性。重要的是,该界面工程策略整合了缺陷工程化正极材料对电解液添加剂和催化中间体的选择性吸附,为催化剂设计与电解液优化耦合建立了一个合理的框架。
尽管取得了这一基础性进展,研究人员认为当前研究侧重于界面机制,是在实验室优化条件下(例如,适中的硫载量和电解液体积)进行的。虽然这些条件有助于分离和验证所提出的S₃∙⁻介导路径,但它们本质上限制了所实现的电池级能量密度。要将这一机制创新转化为高能量密度电池(>300 Wh kg⁻¹)的实际应用,需要同时进行高硫载量正极、贫电解液操作和可扩展电极制造的工程化。然而,即使在当前非优化构型下,所提出的策略已经展现出优异的电化学性能。所设计的锂硫电池在1 C倍率下循环500次后仍保持85.4%的容量,对应每圈仅0.03%的低平均衰减率。作为实际应用的初步验证,它在5 C倍率下还提供了659.6 mAh g⁻¹的高初始比容量,并在4.6 mg cm⁻²的高硫载量下提供了1126.9 mAh g⁻¹的比容量。
除了LSBs性能的直接提升,该研究提出了一个通过自由基介导来调控界面反应路径的通用设计原则。通过控制正极/电解液界面的微溶剂化环境和中间体分布,可以解锁传统醚基体系中无法实现的新催化行为和反应动力学。这些发现为下一代高能可充电电池的发展提供了概念性见解和实践指导,并进一步拓展了界面化学的工具箱。


 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
