北京大学刘文/高艺轩&北京林业大学李璠,最新AFM!轴向氧桥键诱导电子高自旋并破坏铁单原子位点d-π共轭以促进CO₂电还原!孙玮良一作
精细调控催化剂中单原子位点(SAS)电子结构对于提升电催化CO₂还原(CO₂RR)性能至关重要,但仍具挑战性。
2026年01月14日,北京林业大学李璠、北京大学刘文/高艺轩团队合作在Advanced Functional Materials期刊发表题为“Axial O-Bridge Bond Induced Electron High-Spin and Destroyed d–π Conjugation of Fe Single-Atom Sites for Enhanced CO2 Electroreduction”的研究论文,北京林业大学/北京大学孙玮良为论文第一作者,刘文、李璠、高艺轩为论文共同通讯作者。
第一作者:孙玮良
通讯作者:刘文、李璠、高艺轩
通讯单位:北京大学、北京林业大学
论文DOI:10.1002/adfm.202529252
该研究在ZIF-8衍生氮掺杂碳骨架催化剂中构建了轴向氧桥键配位Fe单原子(O─Fe─N₄ SACs),成功实现了Fe 3d轨道自旋调控并破坏了d-π共轭。实验与理论结果表明,轴向O桥键打破了Fe–N₄活性中心的D4h对称性,导致Fe 3d轨道中自旋电子重排,从而破坏了Fe–N–C结构的表面d-π共轭。同时,邻近轴向O桥键的Jahn–Teller效应可有效诱导Fe自旋态由中自旋转变为高自旋。因此,d轨道上成对电子占据数降低对于活化*COOH中间体至关重要,从而显著提升了电催化CO₂RR活性。优化后的催化剂在CO₂RR过程中表现出优异的法拉第效率(FE,-0.7 V下为98.67%),较无轴向配位的Fe–N₄SACs提高了5.6倍。该研究为通过破坏D4h对称性SACs的d-π共轭策略调控电子自旋态以增强电催化CO₂RR性能提供了新见解。
将CO₂电化学转化为碳中性燃料(例如CO和CH₄)是实现碳能源循环的一种前景广阔的策略。在近期开发的CO₂还原反应(CO₂RR)催化剂中,由氮掺杂碳骨架负载的过渡金属催化剂(TM–N─C,M = Fe、Co、Ni等),尤其是对称平面四氮配位单原子催化剂(SACs),已得到广泛研究。然而,在金属单原子(SAS)掺杂N─C骨架中,金属原子与N─C原子共面,由于对称D4h电子分布使得原子轨道平行排列,易于形成d–π共轭效应。这些平行排列的d–π共轭轨道共同构成了扩展的π键网络。然而,过度的d–π共轭可能会限制金属中心的轨道重叠成键与电子传输,最终抑制催化CO₂RR性能。研究表明,轴向配位可有效调控过渡金属的自旋极化并破坏d–π共轭,从而提升CO₂RR活性。
通常,过渡金属易于拥有可变电子和3d自旋轨道,这有助于将电子注入*COOH(吸附在材料表面的氢化CO₂分子,“*”代表材料表面)和中间体的最低未占分子轨道(LUMO),从而赋予其催化CO₂RR潜力。在所有研究最多的过渡金属中,Fe的电子自旋态通常表现出显著的多样性,包括配位结构、电子组态和轨道分裂,这使得其自旋构型易于调控。然而,作为决定电子性质的关键内在因素并对催化剂化学行为起主导影响的Fe的电子自旋态,尚未得到深入研究。如何控制Fe原子的电子自旋态以提升反应活性,仍是一项巨大挑战。
受自旋电子构建与轴向配位体系原理的启发,该研究系统性地开发了一种轴向生长策略,结合ZIF-8模板化合成,构建了轴向氧桥键配位Fe单原子催化剂(O─Fe─N₄ SACs),该催化剂因其Fe的电子自旋态受到调控而在提升催化性能方面极具前景。为阐明d–π共轭的内在机制并实现有效的自旋电子调控,所开发催化剂中五氮配位O─Fe─N₄结构的轴向氧桥键诱导了高自旋电子态。在O─Fe─N₄ SACs的四角锥结构中成功构建高自旋电子态后,决速步(RDS)* + CO₂ + H+ + e– → *COOH得到显著促进,从而提升了电催化CO₂RR的整体性能。具体而言,当O─Fe─N₄ SACs作为阴极应用于电化学CO₂RR体系时,观察到极高的CO转化法拉第效率(FE,在-0.7 V下为98.67%)以及良好的CO₂RR长期耐久性。O─Fe─N₄ SACs的FE值是传统的平面但无轴向氧配位的Fe─N₄ SACs的5.6倍。实验和密度泛函理论DFT计算均表明,轴向氧桥键能够诱导自旋极化从中自旋向高自旋转变,用于填充*COOH的σ*与π*轨道。此外,相邻O─Fe─N₄的高自旋电子需要克服更小的配对能,从而增强了沿轴向的电子传输。此外,Fe─O轴向配位能够更有效地破坏平坦的d–π共轭,并精细调控Fe单原子位点(Fe SAS)的表面自旋态,从而主导表面电化学反应动力学。总体而言,该研究为电化学CO₂转化领域中应用的过渡金属SAS掺杂N─C骨架催化剂的调控与优化提供了新范式。
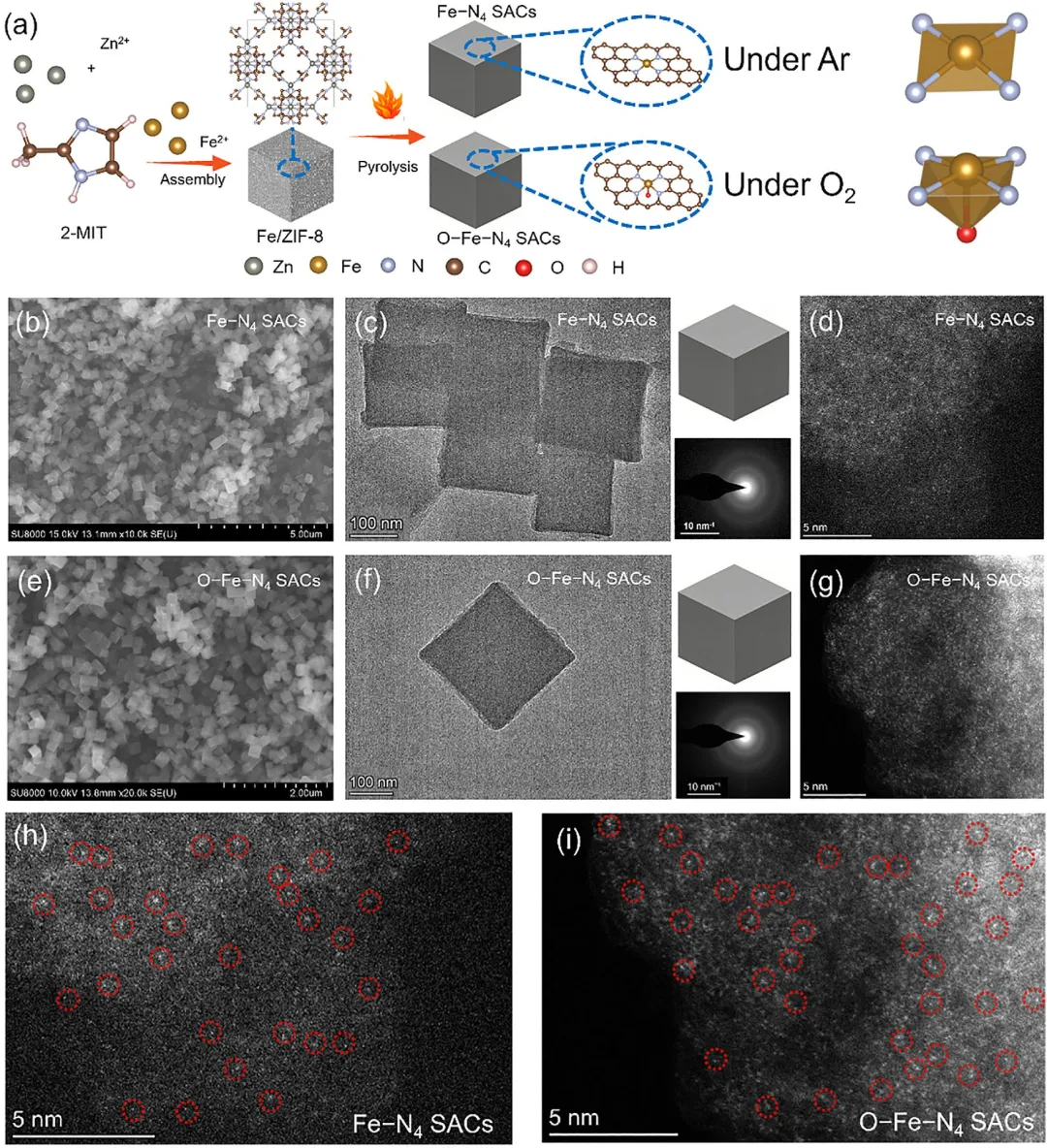
图 1 | 所开发催化剂的合成与表征。(a) Fe–N₄单原子催化剂和O–Fe–N₄单原子催化剂的合成示意图。(b, e) SEM图,(c, f) TEM图(右下角插图显示SAED图),以及(d, g) Fe–N₄单原子催化剂和O–Fe–N₄单原子催化剂的HAADF-AC-STEM图。(h) Fe–N₄和(i) O–Fe–N₄单原子催化剂的放大HAADF-AC-STEM图。
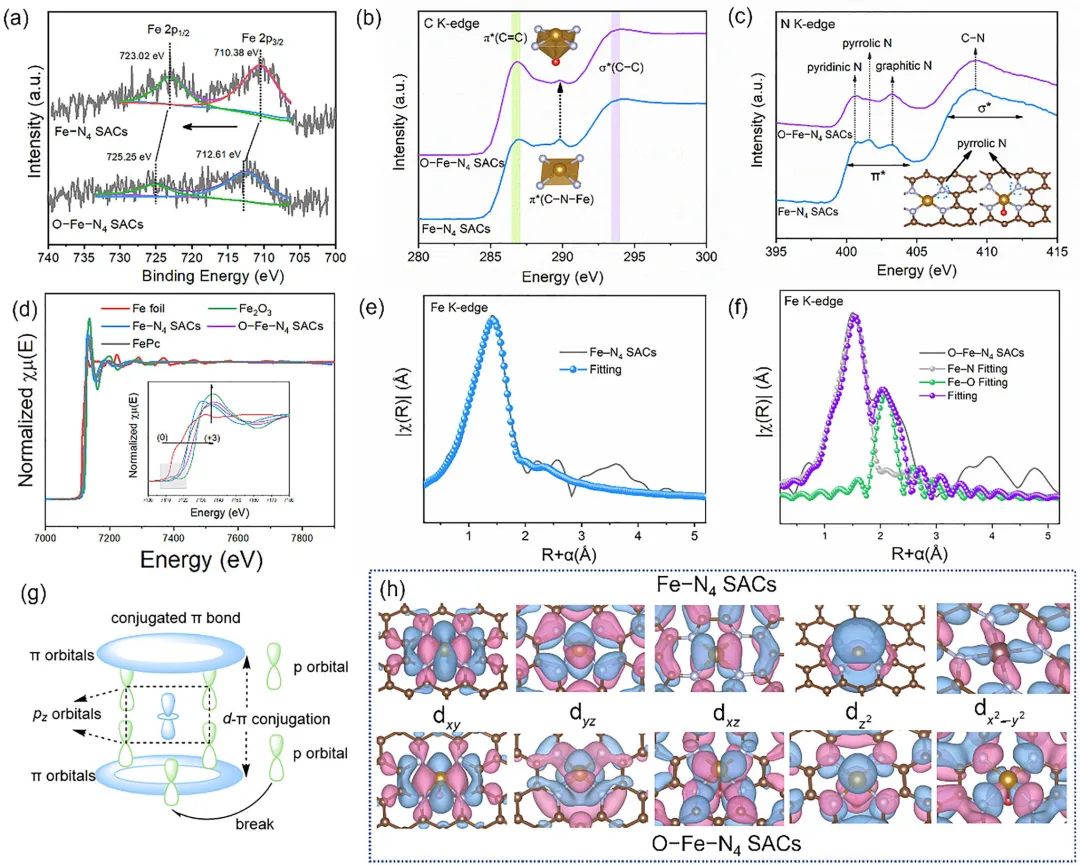
图 2 | 原子结构表征与Fe单原子位点的化学构型。(a) Fe 2p高分辨XPS谱图。(b) C K边和(c) N K边XANES谱图(插图:优化结构模型)。(d) 归一化Fe K边XANES谱图。(e) Fe–N₄单原子催化剂和(f) O–Fe–N₄单原子催化剂的FT k²加权Fe K边EXAFS谱图及相应的EXAFS拟合结果。(g) d-π共轭结构破坏示意图。(h) dxy, dyz, dz², dxz和dₓ²–ᵧ²轨道的波函数。
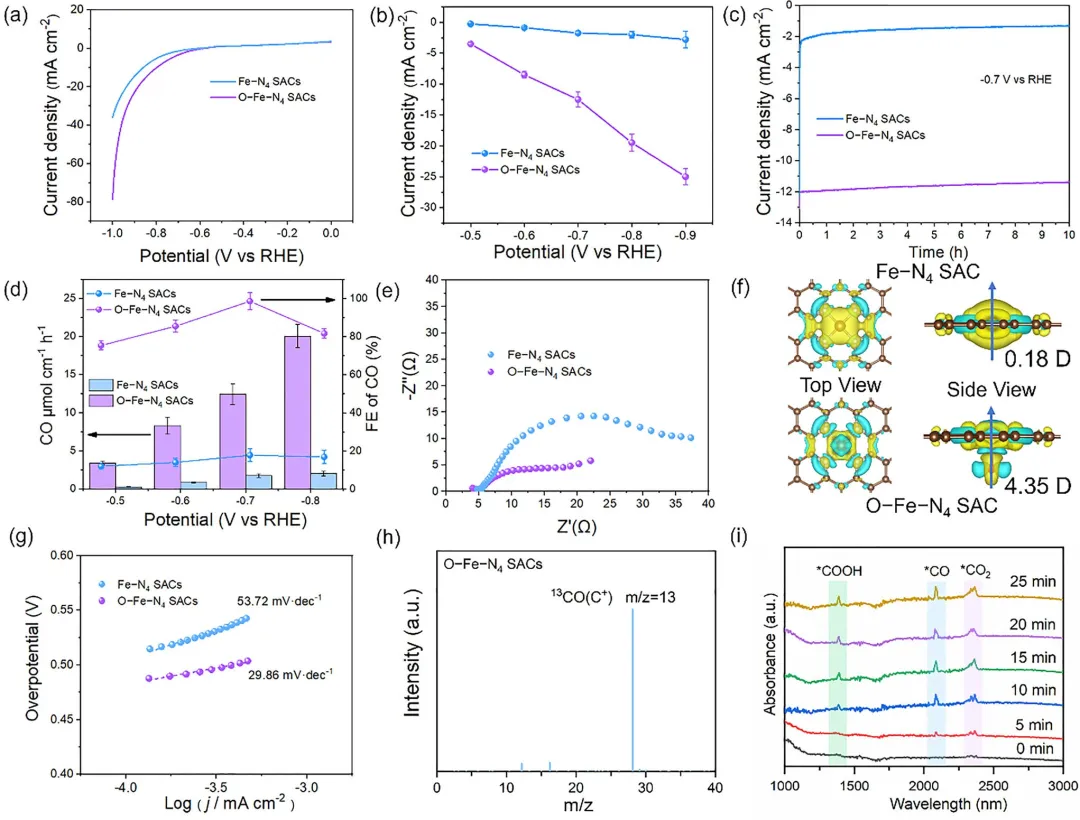
图 3 | CO₂还原的电化学性能。(a) LSV测试,(b) JCO,(c) 长期循环稳定性,(d) FECO,(e) EIS,(f) 电荷密度差,(g) 不同催化剂的Tafel曲线。(h) O–Fe–N₄单原子催化剂的GC-MS对¹³CO产物的分析。(i) CO₂RR过程中O–Fe–N₄单原子催化剂的原位ATR-FTIR分析。(b)和(d)中的数据为三次独立实验的平均值±标准偏差(n=3)。
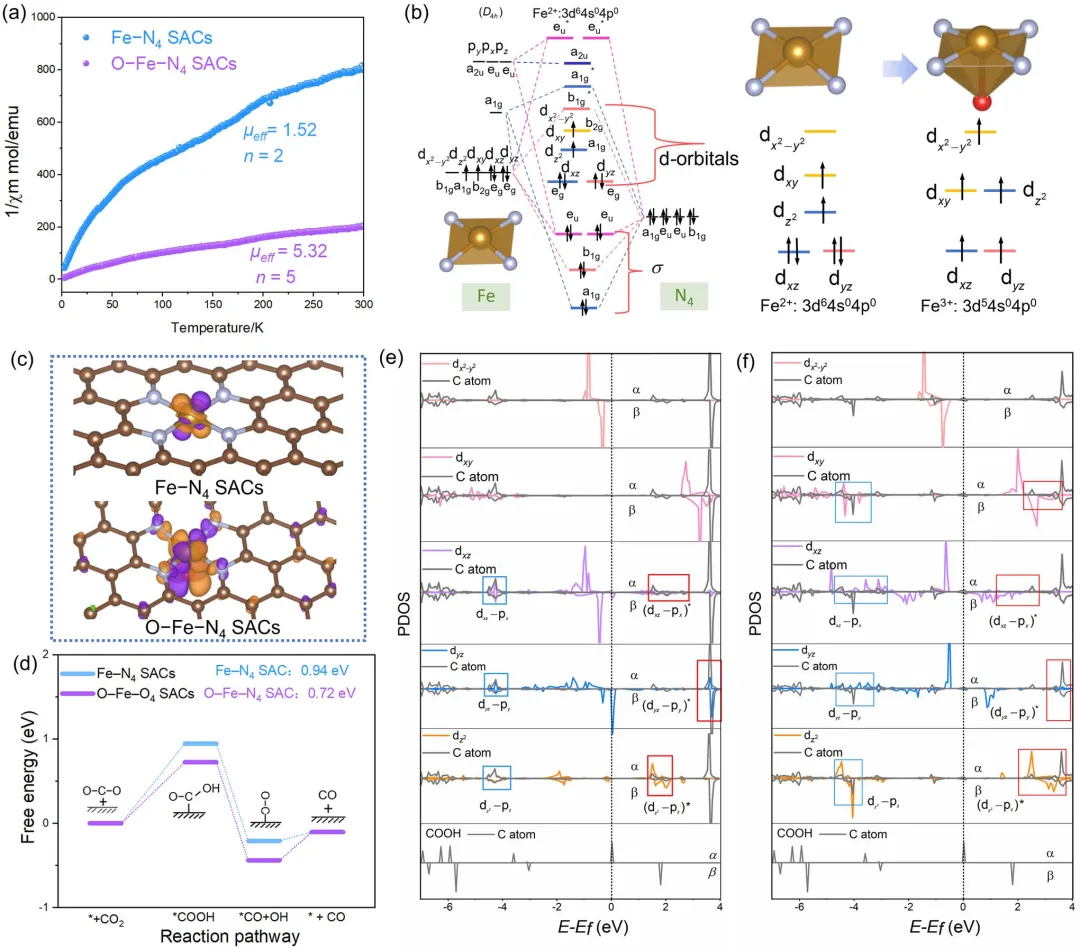
图 4 | 电子自旋态表征、Fe配位构型与DOS谱图。(a) 由M-T测试获得的1/χ图及μeff。(b) Fe–N₄和O–Fe–N₄单原子催化剂的表面自旋电子与群轨道。(c) Fe–N₄和O–Fe–N₄单原子催化剂的自旋电子密度。(d) CO₂RR路径的能量变化曲线。(e) Fe–N₄单原子催化剂和(f) O–Fe–N₄单原子催化剂上Fe单原子位点吸附*COOH的PDOS。
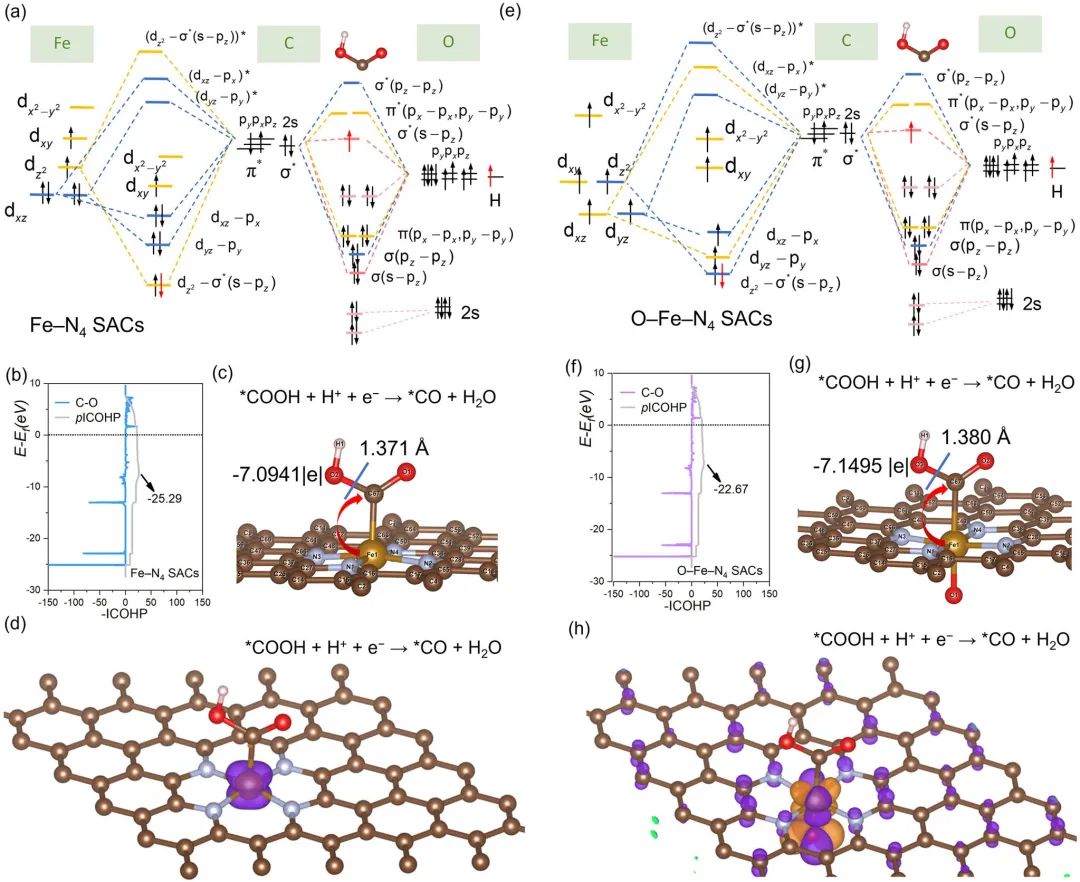
图 5 | Fe–N₄和O–Fe–N₄单原子催化剂中Fe自旋构型与-COOH分子轨道图。吸附COOH的Fe与C原子间成键与反键轨道的示意图(a, e)。(b) Fe–N₄单原子催化剂和(f) O–Fe–N₄单原子催化剂中*COOH的C–OH键的pCOOH与-pCOOH。(c) Fe–N₄单原子催化剂和(g) O–Fe–N₄单原子催化剂中*COOH的Bader电荷与键长。(d) Fe–N₄单原子催化剂和(h) O–Fe–N₄单原子催化剂在吸附COOH后Fe单原子位点的自旋电子密度。
总之,该研究通过轴向生长与ZIF-8模板化策略成功构建了轴向O桥键配位Fe单原子催化剂。轴向O原子有效破坏了平面d-π共轭,并精细调控了Fe单原子位点的表面自旋态,从而主导了表面电化学反应动力学并促进了CO₂RR。相较于平面配位的Fe–N₄单原子催化剂,O–Fe–N₄单原子催化剂的dxz具有更多低自旋电子,dxz与π*(px)之间的强相互作用导致了占据的dxz–π*(px)成键轨道与未占据的(dxz–π*(px))*反键轨道。此外,在CO₂RR过程中,更多的自旋自由电子填充了-COOH的π*轨道,从而削弱了C–OH键并促进了OH解离以生成CO。该研究为理解氮掺杂碳材料上金属单原子催化剂的电子结构与其在电化学反应中的前沿轨道之间的复杂相互作用提供了新见解。



 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
