2.1 材料制备、测序与基因组组装
为探究豆科植物的基因组进化,本研究选取了九种重要的粮食豆类。通过从各物种幼叶中提取高质量的基因组DNA,分别构建了PacBio HiFi长读长文库和Hi-C文库,并对鹰嘴豆额外构建了ONT超长读长文库。利用PacBio Sequel IIe和PromethION平台进行深度测序后,采用hifiasm等工具进行de novo基因组组装,获得初步的contigs。随后,整合Hi-C数据,通过Juicer和3D-DNA等流程对contigs进行挂载、排序和方向矫正,并经Juicebox手动校对,最终获得了所有九个物种染色体级别的的高质量参考基因组。
2.2 基因组注释与比较进化分析
在获得高质量基因组序列的基础上,研究采用了整合策略进行基因组注释。首先,利用LTR_retriever和RepeatMasker等工具对转座元件(TE)进行识别和分类。接着,综合ab initio预测、基于同源物种蛋白序列的同源预测以及利用多组织RNA-seq数据获得的转录本证据,通过EVidenceModeler和PASA等工具整合生成了最终的基因注释集。为进行比较分析,使用OrthoFinder构建了包含12个豆科物种的泛基因组,并在此基础上,利用iqtree和PAML等工具构建了物种系统发育树、估算了分化时间,并借助CAFE5分析了基因家族的扩张与收缩动态。此外,还专门对根瘤发育相关基因及抗病NLR基因家族的进化模式进行了深入剖析。
2. 选择性进化与表观基因组学分析
为揭示豆科作物在驯化过程中的遗传变异,研究整合了四个主要豆科作物(大豆、木豆、鹰嘴豆、豌豆)的野生种和地方品种/栽培种的大规模重测序数据。通过计算群体遗传多样性(π)和分化指数(FST),在全基因组范围筛选受到人工选择的区域,并识别出在多种豆科作物中受趋同选择的关键基因。同时,为了探究基因组扩张与调控机制,对11个豆科物种进行了全基因组重亚硫酸盐测序(MethylC-seq)和ATAC-seq。这些表观组学数据被用于分析DNA甲基化模式、染色质开放区域(OCRs)的分布及其与TEs的关系,并鉴定出非甲基化区域(UMRs)和在部分物种中特有的高甲基化OCRs,从而系统地阐明了TE扩增、基因组大小演化与调控元件进化之间的复杂关系。
3.1 粮食豆类的从头基因组组装
选取九种豆类:为探究豆科植物的遗传多样性与适应性进化,对九种全球广泛消费的粮食豆类进行了从头基因组组装。
优化基因组:尽管这些物种已有基因组序列报道,但部分(如普通菜豆、木豆、鹰嘴豆)主要基于短读长测序,需要进一步优化。
采用三代测序:通过PacBio HiFi和ONT超长读长技术,生成了高质量的测序数据,以构建更为精细和完整的基因组。
组装染色体水平:结合Hi-C染色体构象捕获测序数据,成功将所有物种的基因组组装至染色体水平,基因组大小范围从463.3 Mb到13.0 Gb。
评估组装质量:与现有参考基因组相比,新组装的基因组在连续性和完整性上均表现出优越性,例如,新的鹰嘴豆基因组覆盖度更广,scaffold数量大幅减少。
注释基因元件:基因组注释结果显示,转座元件(TEs)占比为51–92%,共预测出26,180–55,519个基因模型,BUSCO评估的完整度普遍超过99%,并鉴定出数千个新基因。
构建泛基因组:整合本研究的九个基因组以及已发表的大豆、野生大豆和白羽扇豆基因组,构建了包含12个物种的泛基因组,共鉴定出35,389个基因家族。
划分四类基因:根据基因家族在不同物种中的存在频率,将其划分为核心基因(core genes,存在于所有12个物种)、软核心基因(soft-core genes,10-11个物种)、壳基因(shell genes,2-9个物种)和云基因(cloud genes,物种特有)。
分析基因特征:与核心基因相比,非核心基因(包括软核心、壳和云基因)在进化上保守性较低,具有更高的非同义/同义替换率(Ka/Ks)、更多的TEs、更高的DNA甲基化水平以及更低的表达水平。
揭示功能分化:基因本体论(GO)富集分析表明,核心基因主要富集于氮化合物生物合成等基础生命过程,而非核心基因则更多地富集于胁迫响应等与环境适应相关的生物学过程。
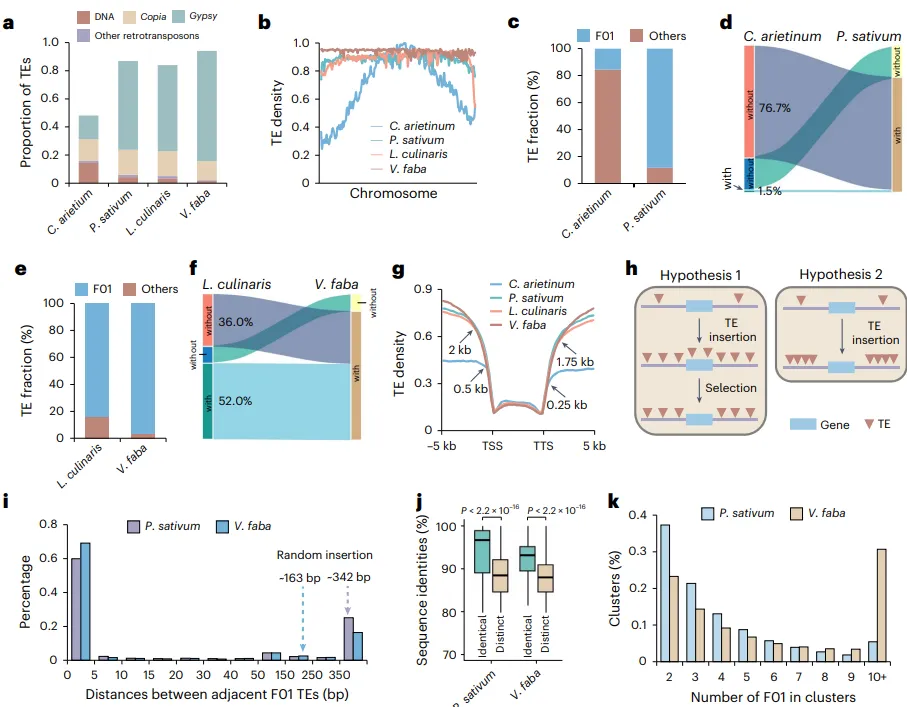
为深入探究豆科植物的遗传多样性和适应性进化,本研究对九种主要的粮食豆类进行了高深度的de novo基因组组装。通过结合PacBio HiFi长读长、ONT超长读长(仅鹰嘴豆)以及Hi-C技术,成功构建了所有物种的染色体级别高质量参考基因组,其质量显著优于多个已发表的版本。基因组注释结果揭示了各物种中基因和转座元件(TEs)的构成情况。在此基础上,通过对12个豆科物种(包含3个已发表基因组)进行泛基因组分析,将所有基因家族划分为核心(core gene sets)、软核心(softcore gene clusters)、壳(shell gene sets)和云(cloud gene clusters)四类。深入分析发现,这四类基因在进化保守性、基因结构、表观遗传修饰及基因功能上表现出显著分化:核心基因功能保守,主要参与基础代谢;而非核心基因(dispensable genes)则演化迅速,富含TEs,并主要参与物种特异性的环境适应过程,如胁迫响应等 (Fig. 1)。
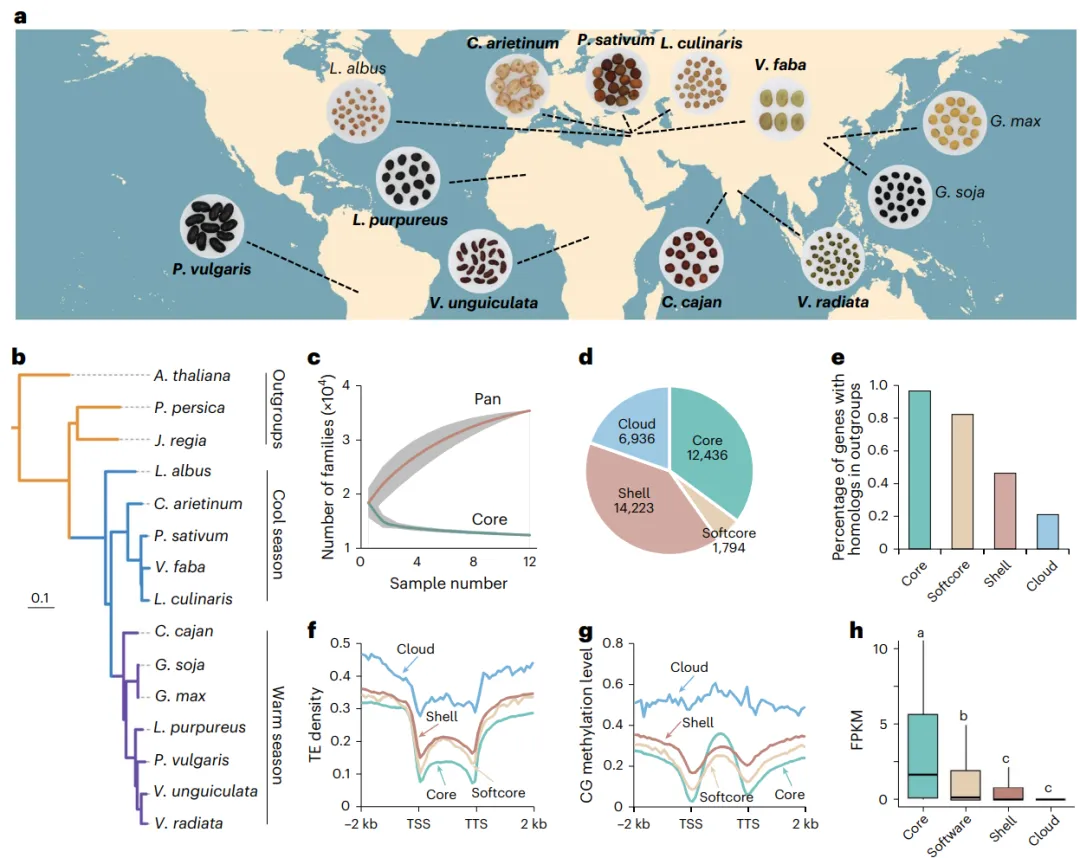
图1. 豆科植物的泛基因组分析。a, 豆科植物的地理分布。粗体字表示本研究中组装的物种,包括普通菜豆(Sucaidou 19-17)、鹰嘴豆(XJ01)、豌豆(Suwandou 08)、兵豆(SX01)、蚕豆(P17-186)、木豆(SM01)、豇豆(Sujiangdou 01)、绿豆(Sulvdou 07)和扁豆(Bianhong 01)。b, 以拟南芥(A. thaliana)、桃(P. persica)和核桃(J. regia)为外群构建的豆科植物最大似然法系统发育树。c, 随豆科植物基因组数量增加,泛基因和核心基因家族数量的变化。数据以平均值±标准差表示。d, 核心、软核心、壳和云基因家族的比例。e, 核心、软核心、壳和云基因中,在外群中存在同源基因的百分比。f, 核心、软核心、壳和云基因周围TEs的分布。g, 核心、软核心、壳和云基因周围CG甲基化的分布。h, 核心基因(30,891个)、软核心基因(3,806个)、壳基因(15,511个)和云基因(572个)的表达水平。每列上方的不同小写字母表示在P < 0.05水平下具有统计学显著性。显著性通过单因素方差分析(ANOVA)检验,多重比较使用Scheffe检验进行分析。箱体的上、下边缘分别代表75%和25%四分位数,中心线表示中位数,须线延伸至1.5倍四分位距(IQR)。TSS,转录起始位点;TTS,转录终止位点。
3.2 根瘤形成相关基因的进化
鉴定根瘤基因:基于已发表的RNA-seq数据,在大豆中鉴定出1,305个根瘤偏好表达基因,其中约54%为核心基因,功能富集于嘌呤生物合成、细胞稳态等。
追溯基因起源:系统发育分析表明,超过70%的根瘤偏好表达基因和超过90%的已克隆根瘤相关基因,其起源早于固氮分支(NFC)的形成,甚至在能形成丛枝菌根(AM)共生的非豆科植物中也存在。
发现新生基因:通过比较基因组学分析,在豆科植物中鉴定出26个de novo(从头起源)基因,它们在所有外群物种中均不存在。其中5个基因在根瘤中高表达,例如编码一个CLAVATA3/ESR相关肽的GmRIC2,该基因参与根瘤数量的负反馈调控。
解析NIN进化:对根瘤形成关键转录因子NIN的系统发育分析发现,凉季豆类中该基因通常为单拷贝,而暖季豆类中则发生了基因复制,存在2-4个拷贝,这可能反映了对不同环境的适应性响应。
对根瘤共生固氮相关基因的进化分析显示,许多关键基因的起源远早于豆科植物与根瘤菌共生关系的建立。在大豆中鉴定出的根瘤偏好表达基因中,大部分为核心基因,且超过70%的基因在非固氮植物中也存在同源基因,表明共生固氮系统可能“借用”了古老的丛枝菌根(AM)共生通路中的组分。更有趣的是,研究发现了26个在豆科植物中de novo起源的基因,其中一个编码调控根瘤数量的肽类激素(GmRIC2),暗示植物可能进化出了新的元件来精细调控共生固氮的平衡。此外,关键转录因子NIN在暖季豆类中发生了谱系特异性的基因复制,而在凉季豆类中则保持单拷贝,这揭示了不同分支豆类在调控共生固氮方面可能采取了不同的进化策略 (Fig. 2)。
图2. 豆科植物中根瘤相关基因的进化。a, 根瘤偏好表达基因中核心、软核心、壳和云基因的比例。b, 带有基因等级的豆科植物系统发育树。G. max中的基因被分配到0-7级。等级0表示G. max中的物种特异性基因。c, 不同基因等级中根瘤偏好表达基因的比例。d, 鉴定豆科植物中de novo基因的流程。e,RIC基因的系统发育分析。f,NIN基因的系统发育树。La, L. albus; Vf, V. faba; Lc, L. culinaris; Ps, P. sativum; Ca, C. arietinum; Mt, M. truncatula; Lj, L. japonicus; Cc, C. cajan; Lp, L. purpureus; Pv, P. vulgaris; Vu, V. unguiculata; Vr, V. radiata; Gm, G. max; Gs, G. soja; Jr, J. regia。
3.3 豆科植物进化过程中的基因扩张与丢失
分析基因家族扩张:凉季和暖季豆类约在5500万年前分化后,各自经历了显著的基因家族扩张,但扩张的基因家族重叠度极低。凉季豆类扩张的基因家族主要富集于光合作用和碳固定,而暖季豆类则富集于防御反应和胁迫响应。
比较抗病基因:作为重要的抗病基因,NLR基因家族在暖季豆类中的拷贝数显著高于凉季豆类,这可能与暖季高温环境下病原体更易滋生有关。
探究基因丢失模式:在经历全基因组复制(WGD)后的二倍化过程中,非核心基因(dispensable genes)相比于核心基因更容易丢失其重复的拷贝。
揭示基因数量增加:在没有近期WGD事件的凉季豆类中,基因数量的增加主要由小规模基因复制(SSD)驱动,且新产生的SSD基因有很高比例起源于远古的WGD基因。
比较基因组学分析揭示,凉季和暖季豆类在约5500万年前分化后,走向了不同的适应性进化道路。两者扩张的基因家族功能迥异:凉季豆类倾向于扩张与光合作用和碳代谢相关的基因,而暖季豆类则显著扩张了与防御、胁迫响应相关的基因,例如其NLR抗病基因的拷贝数远高于凉季豆类。基因丢失模式分析表明,在多倍化后的基因组重塑过程中,功能上非必需的非核心基因更容易丢失。有趣的是,对于没有经历近期全基因组复制的凉季豆类,其基因数量的增加主要依赖于小规模基因复制(SSD),这为物种适应新环境提供了遗传创新的来源 (Fig. 3)。
图3. 豆科植物进化中的基因扩张与丢失。a, 凉季和暖季豆类之间扩张基因家族重叠的维恩图。b,c, 凉季豆类(b)或暖季豆类(c)扩张基因家族中基因的显著富集GO条目。d, 不同豆科物种间NLR拷贝数的比较。e,f,G. max(e)和P. vulgaris(f)中,WGD、SSD和单拷贝基因中核心、软核心、壳和云基因的百分比。g,C. arietinum, P. sativum, L. culinaris 和 V. faba 中WGD、SSD(串联、邻近和散在)和单拷贝基因的数量。h, 在P. sativum(相对于C. arietinum)和V. faba(相对于L. culinaris)中,新产生的SSD基因来源于WGD基因的百分比。基因组中的其他基因用作对照。统计显著性通过双侧Fisher精确检验确定。TNL,至少含一个Toll/interleukin-1受体(TIR)结构域的TIR–NLR基因;CNL,同时含有CC和NB-ARC结构域的CC–NLR基因;RNL,至少含一个RPW8结构域的CCR–NLR基因;NL,仅含NB和LRR的NLR基因。
3.4 豆科植物进化中的趋同选择
扫描趋同选择:通过比较大豆、木豆、鹰嘴豆和豌豆的野生种与地方品种/栽培种的基因组,在全基因组范围内扫描因人工选择而导致遗传多样性降低的区域。
鉴定选择基因:共鉴定出226个基因在上述四种豆科作物中表现出趋同选择的信号,这些基因主要参与种子休眠、种子增大和能量平衡等途径。
分析关键基因:一个典型的例子是GmYUC4a及其直系同源基因,它们在四种作物中均受到选择。该基因编码一个生长素合成酶,与种子大小正相关。
验证基因功能:通过对GmYUC4a的单倍型分析发现,栽培种中频率显著增加的单倍型(Hap2/Hap3)与更高的种子重量相关。利用染色体片段替换系(CSSLs)进行的实验验证也证实,将野生大豆的YUC4a等位基因(Hap1)导入栽培种后,会导致籽粒变小、百粒重显著降低。
为探究不同豆科作物在独立驯化过程中是否存在平行的遗传改良模式,研究对大豆、木豆、鹰嘴豆和豌豆进行了全基因组范围的选择信号扫描。结果发现,共有226个基因在这些物种中均表现出遗传多样性显著降低的趋同选择特征,这些基因功能上与种子休眠和种子大小等关键农艺性状相关。其中,一个编码生长素合成关键酶的基因YUC4在四个物种中均受到强烈选择。对大豆中GmYUC4a的单倍型分析和功能验证实验均证实,在驯化过程中被偏好选择的等位基因能够显著提高基因表达水平,从而增加种子重量。这一发现表明,尽管豆科作物在不同地区被独立驯化,但它们可能通过选择相同的保守基因来改良相似的农艺性状 (Fig. 4)。
图4. 豆科植物进化过程中的趋同选择。a, 大豆、木豆、鹰嘴豆和豌豆中,野生种与地方品种之间的全基因组π比率。πW表示野生大豆的π值,πL表示地方品种的π值。b, 大豆中受人工选择的基因数量。1,仅在大豆中受选择的基因;2和3,分别在大豆和另外一个或两个物种(木豆、鹰嘴豆、豌豆)中受趋同选择的基因;4,在所有四个物种中均受趋同选择的基因。c, 在大豆、木豆、鹰嘴豆和豌豆中受趋同选择的基因的共线性关系。d, 大豆中GmYUC4a(GmW82.14G124100)及其在木豆(CcYUC4, Cajca.01G068800)、鹰嘴豆(CaYUC4, Cicar.07G377300)和豌豆(PsYUC4, Pissa.06G068100)中直系同源基因的600 kb侧翼基因组区域内,野生种与地方品种间的π比率。π比率使用100 kb滑动窗口,步长10 kb计算。e, 大豆中GmYUC4a的单倍型分析。f, 携带Hap1(n=6)、Hap2(n=807)和Hap3(n=56)的材料的百粒重。P值由双尾学生t检验确定。箱体的上、下边缘分别代表75%和25%四分位数,中心线表示中位数,须线延伸至1.5倍IQR。g, 中国栽培品种SN14及其两个独立的染色体片段替换系(R3和R170)的代表性种子。h, SN14、R3和R170的百粒重。数据以平均值±标准差表示,每组五个生物学重复。P值由双尾学生t检验确定。SN14,绥农14。
3.5 转座元件的局部增殖导致基因组扩张
分析TE扩张模式:与暖季豆类相比,凉季豆类的基因组扩张更为显著,主要由TEs在常染色质区的扩增驱动,特别是Gypsy超家族的LTR反转录转座子。
鉴定关键TE家族:通过比较亲缘关系相近但基因组大小差异巨大的物种对(如鹰嘴豆/豌豆,兵豆/蚕豆),发现一个名为F01的Ogre类TE家族是驱动基因组扩张的主要动力。
揭示TE增殖机制:F01 TEs的扩张呈现两步模式:在从鹰嘴豆到豌豆的进化中,主要通过de novo插入到新位点;而在从兵豆到蚕豆的进化中,则主要通过对已存在F01元件的再次扩增。
提出串联扩增假说:TEs的扩增并非随机插入,而是倾向于在基因贫乏区以“簇状”或“串联”的方式插入,相邻TEs之间的距离极短。这一现象支持了TEs通过识别邻近基序进行靶向插入的“串联扩增”假说,而非随机插入后被选择性清除。
对豆科植物基因组大小演化的研究发现,TE扩增是主要驱动力,尤其是在凉季豆类中。基因组扩张主要发生在常染色质区,由Gypsy超家族的LTR反转录转座子(特别是F01家族,即Ogre元件)驱动。比较基因组学分析揭示了一种两阶段的扩增模式:在基因组相对较小的物种向中等大小物种演化时(如鹰嘴豆到豌豆),TEs主要通过de novo插入基因间隔区;而在基因组进一步扩张时(如兵豆到蚕豆),则表现为对已有TEs的再次扩增。深入分析TE插入位点的分布模式,发现TEs并非随机插入,而是倾向于在基因稀疏的区域形成紧密的簇,相邻TEs间距极小。这一发现强烈支持了“串联扩增”假说,即TEs可能通过识别特定基序实现局部、集中的增殖,从而高效地扩大基因组,同时避免破坏基因功能区 (Fig. 5)。
图5. 豆科植物进化过程中TEs的扩张。a, 常染色质区中TEs的比例。b, TEs在整个染色体上的分布。x轴代表从起始到末端的染色体坐标。c, 豌豆和鹰嘴豆的Gypsy元件中F01 TEs的百分比。分析使用了豌豆和鹰嘴豆之间两个连续基因的共线性区域。d, 鹰嘴豆和豌豆中含有或不含F01 TEs的共线性区域的比例。e, 兵豆和蚕豆的Gypsy元件中F01 TEs的百分比。分析使用了兵豆和蚕豆之间两个连续基因的共线性区域。f, 兵豆和蚕豆中含有或不含F01 TEs的共线性区域的比例。g, 四种凉季豆类保守基因周围TEs的分布。h, 豆科植物进化过程中TE扩张的两种假说。假说1表示TEs随机插入基因组。假说2表示TEs以具有插入位点偏好性的方式成簇插入。i, 豌豆和蚕豆中相邻F01 TEs之间距离的分布。j, 在相同区域(豌豆n=48,173;蚕豆n=328,793)或不同区域(豌豆n=9,676,037;蚕豆n=57,915,589)的F01 TEs之间的序列一致性。P值通过双侧Wilcoxon秩和检验计算。箱体的上、下边缘分别代表75%和25%四分位数,中心线表示中位数,须线延伸至1.5倍IQR。k, 含有不同数量F01 TEs的簇的百分比。间距不超过5 bp的相邻TEs被聚合成一个簇。
3.6 调控元件的进化
分析TE衍生调控区:TEs可以演化为调控元件,形成开放染色质区域(OCRs)。随着基因组增大,由TE完整序列构成的OCRs(cTE-OCRs)比例从鹰嘴豆的0.8%增加到蚕豆的10%。
发现高甲基化OCRs:与玉米等植物中OCRs普遍低甲基化的认知不同,在豌豆、兵豆和蚕豆等大基因组豆科植物中,cTE-OCRs表现出异常高的DNA甲基化水平,且其开放染色质的形成不伴随DNA甲基化的降低。
鉴定新型调控元件:除了位于非甲基化区域的常规OCRs(UMR-OCRs),在基因组较大的凉季豆类中还鉴定出大量位于高CG甲基化区域的新型OCRs(CG-OCRs)。
探究形成机制:这些CG-OCRs主要分布在基因远端区域,富含Mutator等DNA转座子,并表现出显著的转录活性和染色质可及性。同时,多个染色质调控相关基因的表达水平随基因组增大而上调,这可能与高甲基化OCRs的形成和功能维持有关。
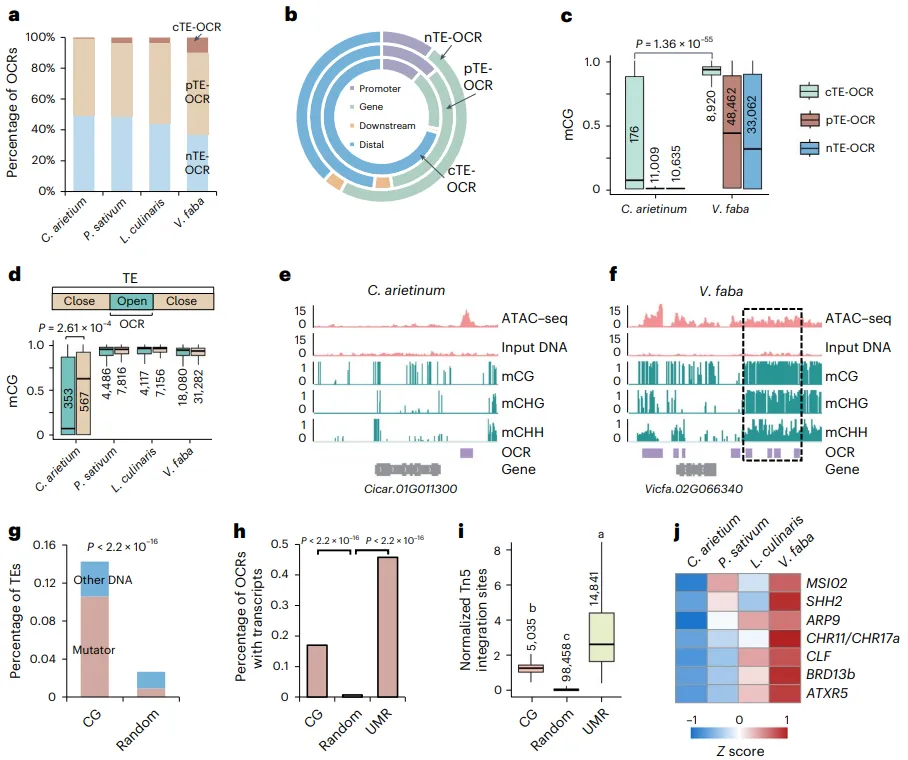
TEs不仅驱动基因组扩张,还能演化为调控元件。分析发现,在基因组较大的凉季豆类中,由TE直接形成的开放染色质区域(cTE-OCRs)比例显著增加。一个反常的现象是,这些物种中的cTE-OCRs及其周围区域表现出极高的DNA甲基化水平,这与OCRs通常位于低甲基化区域的普遍认知相悖。进一步分析鉴定出一类新型的、位于高CG甲基化环境下的功能性OCRs(CG-OCRs),它们在基因组较大的豆科植物中大量出现,主要富集在基因远端的Mutator类DNA转座子区域,并具有显著的转录活性。与此同时,多个染色质重塑相关基因的表达量在这些物种中协同上调。这些结果共同揭示了一种新的调控元件进化模式:在TE驱动的基因组扩张过程中,植物可能进化出了新的表观遗传机制,允许高甲基化的TE区域作为功能性调控元件发挥作用 (Fig. 6)。
图6. 调控元件的进化。a, cTE-OCRs、pTE-OCRs和nTE-OCRs的百分比。b, 蚕豆中基因、启动子(TSS上游2 kb)、下游(TTS下游500 bp)和远端基因间隔区的cTE-OCRs、pTE-OCRs和nTE-OCRs的百分比。c, 鹰嘴豆和蚕豆中cTE-OCRs、pTE-OCRs和nTE-OCRs的DNA甲基化水平。d, TEs内部OCRs(开放)及其余部分(封闭)的CG甲基化水平。对于c和d,数字代表样本量,显著性通过双侧Wilcoxon秩和检验进行测试。e,f, 具有代表性的IGV快照,显示了鹰嘴豆(e)和蚕豆(f)中存在或不存在DNA甲基化的OCRs。g, 蚕豆中与远端CG-OCRs重叠的TEs中DNA转座子的百分比。随机选择的具有CG甲基化的远端区域用作对照。h, 蚕豆中具有转录本的CG-OCRs和UMR-OCRs的百分比。对于g和h,P值由双尾Fisher精确检验确定。i, 蚕豆中CG-OCRs和UMR-OCRs的染色质可及性。数字代表样本量。显著性通过单因素ANOVA检验测试,多重比较使用Scheffe检验分析。j, 在鹰嘴豆、豌豆、兵豆和蚕豆中,参与染色质可及性调控的基因的表达水平变化。在c, d 和 i中,箱体的上、下边缘分别代表75%和25%四分位数,中心线表示中位数,须线延伸至1.5倍IQR。
构建了覆盖广泛、高质量的豆科植物泛基因组资源:研究首次对九种全球重要粮食豆类进行了染色体级别的de novo基因组组装,并结合已有数据构建了包含12个物种的泛基因组。这一高质量、大规模的基因组资源库是前所未有的,为深入解析豆科植物的进化、驯化和农艺性状遗传基础提供了坚实的数据基石。
系统性揭示了豆科作物驯化过程中的趋同选择规律:通过对大豆、木豆、鹰嘴豆和豌豆四种独立驯化的豆科作物进行全基因组选择信号扫描,研究首次在多物种间系统性地鉴定出226个受到趋同选择的基因。并以YUC4基因为例,通过单倍型分析和功能实验证实了其在不同豆科作物中平行地调控种子大小。这为理解作物驯化的共性分子机制和利用趋同进化规律进行育种改良提供了新视角。
创新性地提出了TEs驱动基因组扩张的“串联扩增”模型:针对凉季豆类基因组急剧扩张的现象,研究发现其主要由特定TE家族(F01 Ogre元件)驱动。更重要的是,研究揭示了这些TEs并非随机插入,而是倾向于在基因贫乏区以“簇状”或“串联”的方式密集插入。基于此,研究提出了一个全新的“串联扩增”(tandem amplification)假说来解释TEs的增殖机制,挑战了传统的随机插入-选择清除模型。
发现了与传统认知相悖的高甲基化功能性调控元件:研究发现在豌豆、蚕豆等大基因组豆科植物中,大量开放染色质区域(OCRs)存在于高度DNA甲基化的TEs区域内,这些区域被称为CG-OCRs。这颠覆了“功能性调控元件通常是低甲基化的”传统认知。研究证实这些高甲基化OCRs具有显著的转录活性,代表了一种在基因组扩张过程中演化出的新型调控机制。
首次鉴定出在豆科植物共生固氮系统进化中“从头起源”的关键调控基因:研究通过精细的比较基因组学分析,筛选出26个在豆科植物中de novo起源的基因。其中,GmRIC2被证实为在根瘤中高表达,并参与根瘤数量的负反馈调控。这一发现为理解复杂生物学过程(如共生固氮)如何通过产生全新基因元件来进行精细调控和优化提供了直接证据。
审核|林蘖亙
阅读科研文献,荟萃前沿进展
欢迎关注、转发、投稿、点赞