
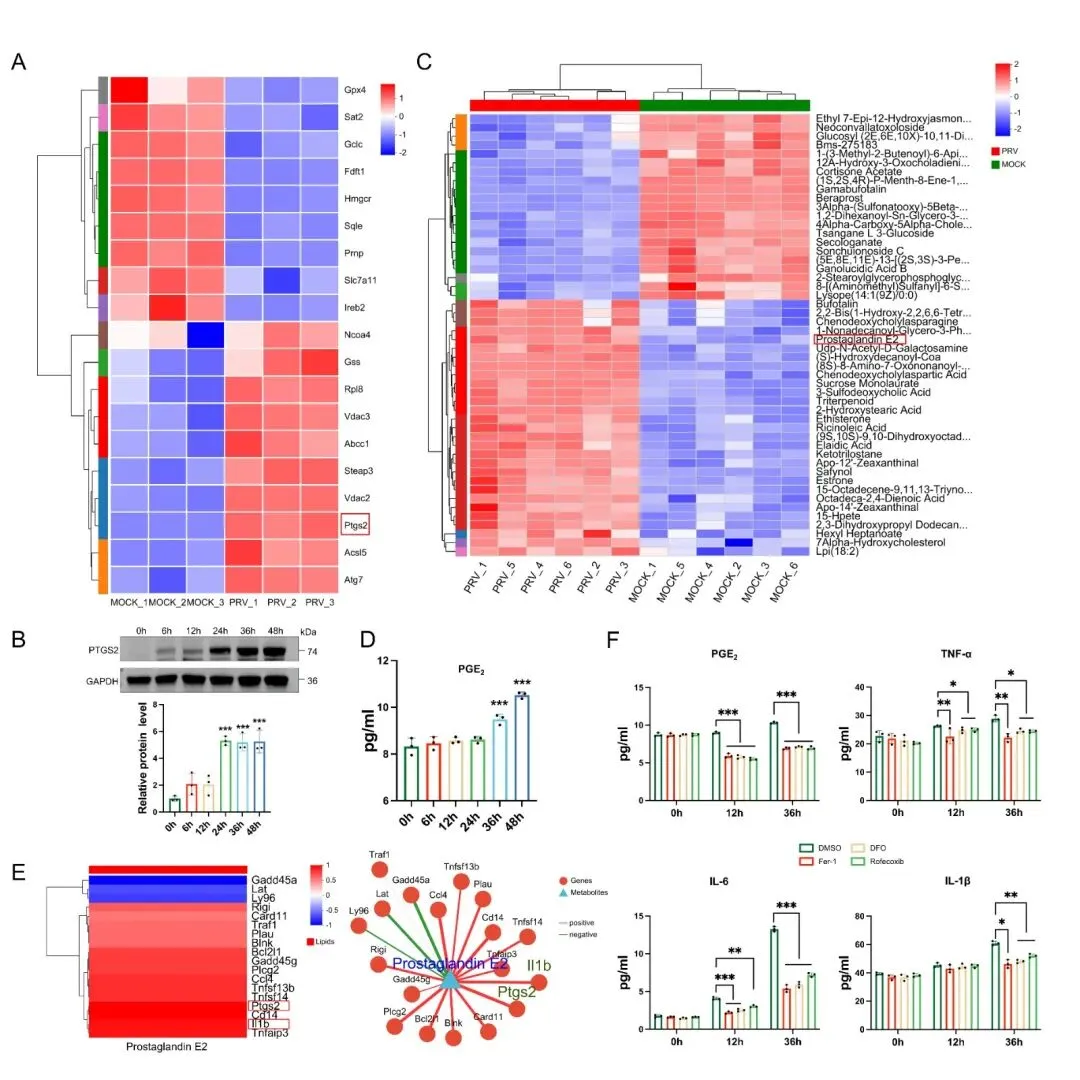
该研究围绕“铁蛋白自噬-铁死亡-神经炎症”调控轴,系统解析了 PRV 感染导致脑损伤的分子机制。研究发现,PRV 感染可在 N2a 细胞、小鼠原代神经元以及小鼠脑组织中诱导铁死亡;PRV 感染能够激活铁蛋白自噬,促进细胞内游离铁水平升高,进而诱发脂质过氧化,最终驱动铁死亡发生;通过转录组学与代谢组学联合分析证实,PRV 感染诱导的铁死亡可通过 PTGS2/PGE2 通路促进神经炎症发生(图1)。
图1. 铁死亡激活 PTGS2/PGE2 通路促进 PRV 诱导的神经炎症反应
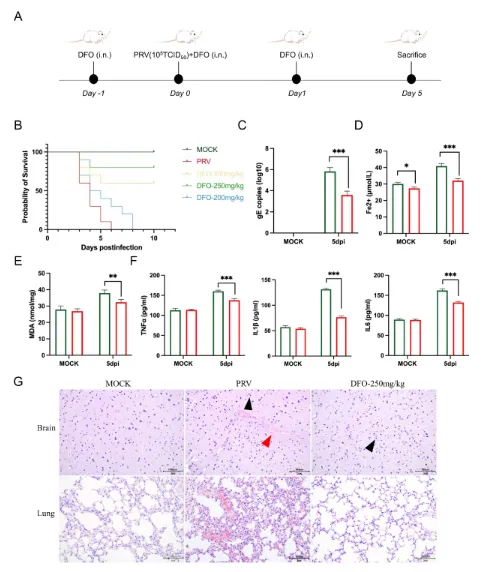
PRV 感染小鼠脑组织中游离铁水平和脂质过氧化程度明显升高,并伴随显著的神经炎症反应和脑组织病理损伤。铁死亡抑制剂去铁胺(DFO)可有效缓解 PRV 感染诱导的铁死亡,降低小鼠脑组织中的病毒滴度,并减轻病毒性脑炎相关病理损伤(图2)。
图2. 铁死亡抑制剂 DFO 对 PRV 感染小鼠的治疗作用
该研究从铁代谢异常与程序性细胞死亡交叉调控的角度,揭示了 PRV 感染引发中枢神经系统损伤的新机制,明确了铁蛋白自噬介导的铁死亡及 PTGS2/PGE2 炎症通路在病毒性脑炎中的重要作用,为 PRV 相关神经系统疾病的防控和治疗提供了新的理论基础与潜在靶点(图3)。
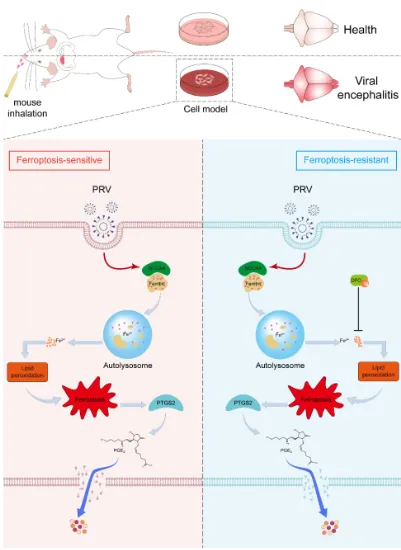
图3. 铁死亡介导 PRV 诱导脑炎机制示意图
中国农业大学动物医学院在读博士研究生孙佳丽为论文第一作者,胡艳欣教授和杨汉春教授为论文通讯作者。该研究得到国家自然科学基金重点项目(32330106)、国家自然科学基金面上项目(32372966, 32172829)、国家生猪产业体系专项基金(CARS-35)和中国农业大学“2115人才培养计划”等项目的资助。

 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
