从基因缺陷到不育表型
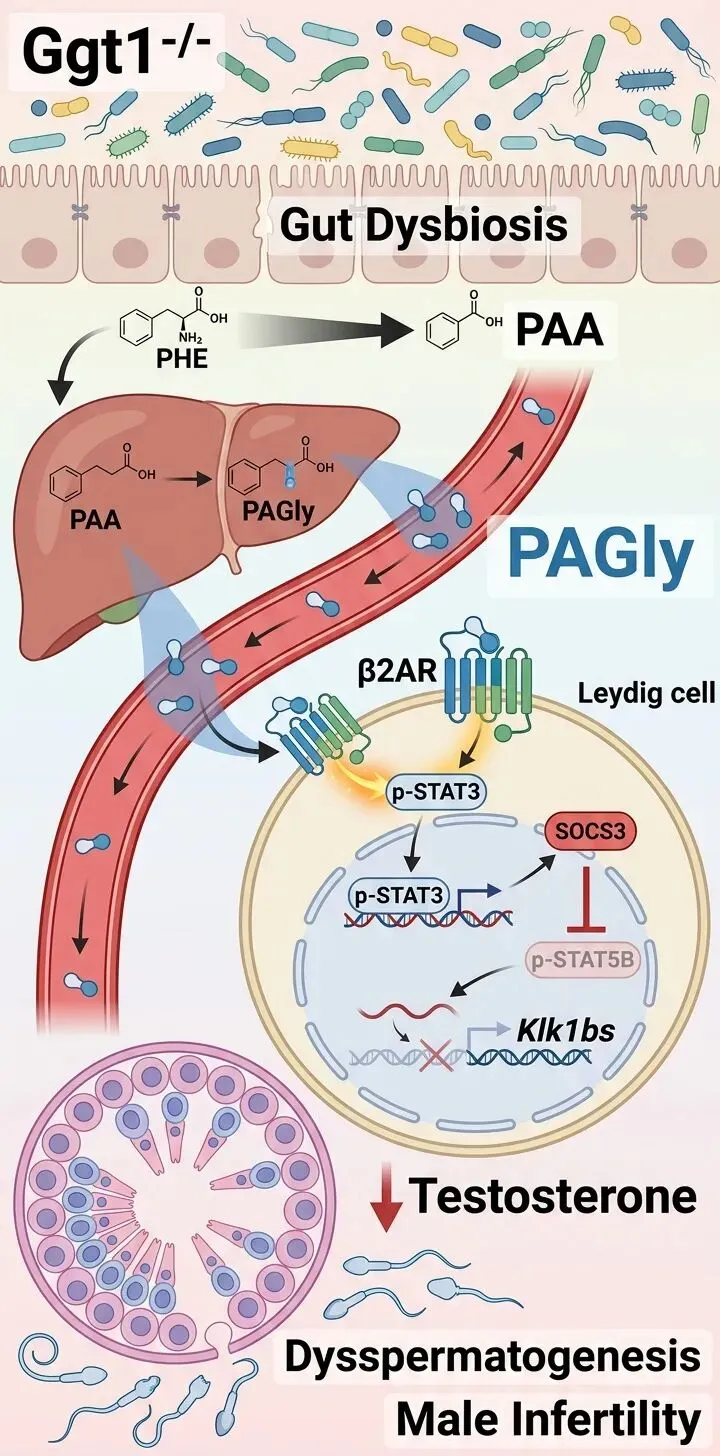
研究始于一个关键的宿主基因——γ-谷氨酰转移酶1(Ggt1)。当研究人员构建了全身性敲除该基因的小鼠模型后,一个引人注目的现象出现了:雄性小鼠完全不育。它们的精子浓度和活力显著下降,异常精子比例激增,睾丸中生精小管结构松散,内部的生殖细胞和睾酮合成关键细胞——间质细胞数量锐减。血液和睾丸内的睾酮水平也随之降低。
然而,深入分析发现,Ggt1基因本身在睾丸组织中几乎不表达。这意味着,敲除该基因对精子发生的破坏,并非由其直接调控睾丸细胞所致,暗示背后存在一条非遗传性的、“迂回”的作用路径。
代谢组学追踪“罪魁祸首”
为了寻找真正的“破坏者”,团队对突变小鼠的血清和睾丸进行了全面的代谢组学分析。在众多发生变化的代谢物中,一个名为苯乙酰甘氨酸(PAGly)的分子引起了注意。它在突变小鼠的血清和睾丸中均异常升高。进一步分析显示,睾丸内PAGly的水平与睾酮含量、精子浓度呈显著负相关。
这个关联是偶然还是因果?为了验证,研究团队直接向正常雄性小鼠腹腔注射PAGly。结果令人震惊:仅注射10天,这些小鼠便重现了基因敲除鼠的不育表型——精子减少、畸形率上升、睾丸结构受损、睾酮合成关键蛋白表达下降。相反,如果用药物(普萘洛尔)阻断PAGly的作用,则可以部分逆转基因敲除鼠的精子发生障碍。这些证据链条表明,PAGly是导致精子发生障碍的关键介质。
菌群失调是代谢异常的源头
PAGly从何而来?它的生物合成路径指向了肠道菌群:膳食中的苯丙氨酸(PHE)首先被肠道细菌代谢为苯乙酸(PAA),后者在宿主肝脏中与甘氨酸结合,最终形成PAGly。那么,Ggt1敲除是否扰动了这条路径?
答案是肯定的。对小鼠粪便的分析显示,突变小鼠肠道中PHE减少,而其下游产物PAA显著增多。宏基因组测序进一步揭示,突变小鼠的肠道菌群结构发生了显著改变,其中能够将PHE转化为PAA的细菌及其相关酶(PPDC和PPFOR)的丰度异常升高。这清晰地表明,宿主基因缺陷导致了肠道菌群失调,进而过度生产了PAGly的前体物质。
最具说服力的证据来自粪菌移植实验。将突变小鼠的肠道菌群移植给经过抗生素处理的“伪无菌”小鼠后,接受移植的小鼠不仅血清和睾丸中PAGly水平升高,还完整地“复制”了供体小鼠的不育表型:精子质量下降、睾丸结构紊乱、激素水平失衡。这直接证明了,失调的肠道菌群足以通过其代谢产物导致生殖功能障碍。
揭示完整的信号传导链
PAGly如何具体损害睾丸功能?随后的分子机制研究描绘出一条清晰的信号通路。研究人员发现,PAGly通过激活睾丸间质细胞表面的β2肾上腺素能受体(β2AR) 发挥作用——这与结构相似的代谢物苯乙酰谷氨酰胺(PAGln)的作用机制相似。
受体激活后,下游的信号转导与转录激活因子3(STAT3)被磷酸化。活化的p-STAT3上调了细胞因子信号抑制因子3(SOCS3)的表达。而SOCS3作为一个关键的负反馈调节因子,抑制了另一个转录因子STAT5B的磷酸化。
这一抑制环节至关重要。团队通过CUT&Tag和ATAC-Seq等表观遗传学分析证实,磷酸化的STAT5B(p-STAT5B)能够结合到一组名为Klk1bs(激肽释放酶家族成员,包括Klk1b21、Klk1b22等)基因的启动子附近,并正向调控其转录。
Klk1bs基因在间质细胞中特异性高表达,对维持正常的睾酮合成至关重要。当PAGly通过β2AR-SOCS3轴抑制了STAT5B的活性后,Klk1bs的表达随之下降,进而导致睾酮合成酶(如CYP11A1、HSD3B1)表达减少,最终瓦解了支持精子发生所必需的激素微环境。
为“肠-睾轴”理论奠定基石
这项研究系统性地阐释了“肠道菌群-代谢物-睾丸”轴在调节雄性生殖健康中的具体作用机制。它首次将宿主基因缺陷、肠道菌群失调、特定代谢物累积、细胞受体信号传导以及睾丸内关键基因的转录调控串联成一个完整的病理模型。
这一发现不仅加深了对男性不育复杂病因的理解,也为未来的临床干预提供了新的思路。监测血液中PAGly水平或许能成为评估男性生殖风险的一个潜在生物标志物,而针对肠道菌群、β2AR信号通路或STAT5B活性的调控策略,则可能为治疗由菌群失衡引起的男性不育症开辟新的途径。
生命体内部不同系统间通过代谢物进行的“远程对话”,其精细与复杂程度远超想象,而这项研究为我们理解这种对话如何影响核心生理功能,提供了关键的一章。










 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
