1.
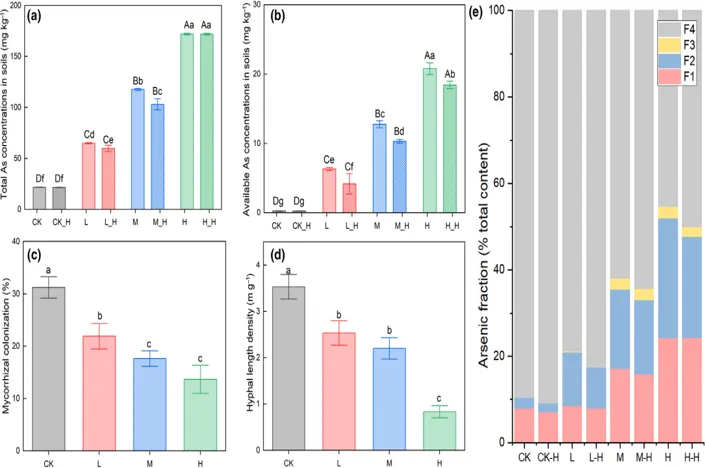
fig.1
(a)土壤中总砷( T-As ) 含量 (b)有效砷( A-As )含量。在相同As浓度下,菌丝际和非菌丝际土壤之间的小写字母表示显著差异,而大写字母表示4个处理之间的显著差异。
(c)寄主植物菌根侵染率 (d)土壤菌丝长度密度。均来自田间菌丝际土壤采集装置。
( e )不同处理土壤As形态的连续提取( BCR法)。后缀_ H表示菌丝际土壤,无后缀表示非菌丝际土壤。
田间菌丝际土壤在三种污染水平下均表现出较低的有效态砷含量,同时菌丝际土壤在BCR的分级中F1与F2两类易迁移形态上也始终低于非菌丝际土壤,这说明AMF菌丝可能通过直接吸附、胞壁结合砷或通过选择性富集微生物来改变微环境来降低砷的活性。
另一方面,随着砷浓度升高,菌根侵染率与菌丝长度密度显著下降,表明砷胁迫会强烈抑制真菌定殖,直接吸附能力受到了限制。然而,在菌丝长度减少的同时,菌丝际中仍能看到有效态砷和易迁移砷形态的下降,提示微生物的间接作用可能在高砷背景下占据更重要地位。
2.
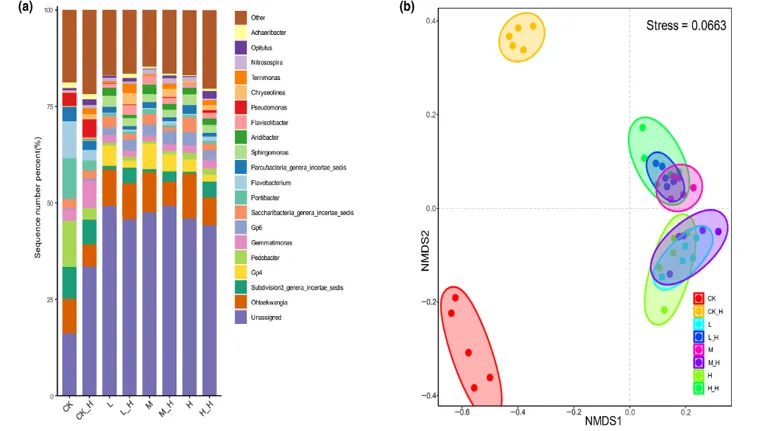
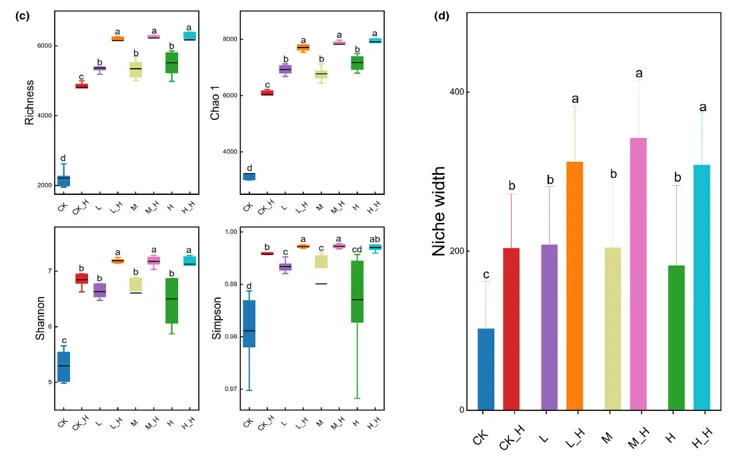
fig.2
( a )不同As污染水平下菌丝际和非菌丝际土壤中前20个细菌属的相对丰度
( b )基于Bray - Curtis距离的非度量多维标度方法( NMDS )
( c )四种alpha多样性指数
( d )生态位宽度的差异。
在砷污染土壤中,随着砷污染浓度增高,菌丝际土壤微生物的α多样性始终显著高于非菌丝际土壤,四种α多样性均稳定提升,表明菌根圈微生物群落展现出显著高于非菌根圈土壤的多样性。NMDS 辅以分析,四个砷水平下菌丝际与非菌丝际样品始终被分为两个分明的群落簇,且菌丝际群落间的离散度明显更低,揭示了菌根圈与非菌根圈土壤微生物群落之间存在显著的结构差异,同时菌根圈微生物群落展现出更宽广的生态位宽度(可能是由于AMF 菌丝分泌的碳源扩大微生物生态位),凸显了AMF菌丝网络在塑造微生物生态位中的重要性。
3.
fig.3
协惯量分析法( CIA )表示丛枝菌根真菌( AMF )与菌丝际细菌群落的关系( RV = 0 . 764 (当RV趋近于1时,表明两个群落之间具有较强的协同相关性)),P = 0。01 )。
箭头代表了两个群落中样本的位置变化。箭头的基部表示样品在AM真菌群落中的位置,而箭头的尖端表示样品在菌丝际细菌群落中的位置。箭头的长度代表了两个群落之间的差异。箭头方向表示样本在两个群落中的位置关系
协惯量分析(CIA)分析表明,在砷污染条件下,AM真菌群落与菌根圈微生物组之间存在一致的协调响应。特别是在高砷浓度下,AM真菌与菌根圈微生物组之间协同变化的一致性得到增强。
关于细菌群落结构和多样性的研究结果表明,AMF菌丝与特定微生物之间已建立共生关系,这是对砷污染的适应性反应。
这种共生关系可能涉及菌丝体提供的营养物质以及微生物在砷转化过程中的参与。菌根圈微生物组可能通过菌丝网络提供的独特土壤微环境,在砷污染管理中发挥重要的动态调控作用。
这些发现支持了第一个假设:砷污染以不同于非菌根圈微生物组的方式重塑菌根际微生物组。随着砷浓度升高,微生物组差异性增加的现象凸显了AM真菌菌丝界面在胁迫下的选择性影响。
4.As污染下核心菌丝际微生物组及其功能关联的概述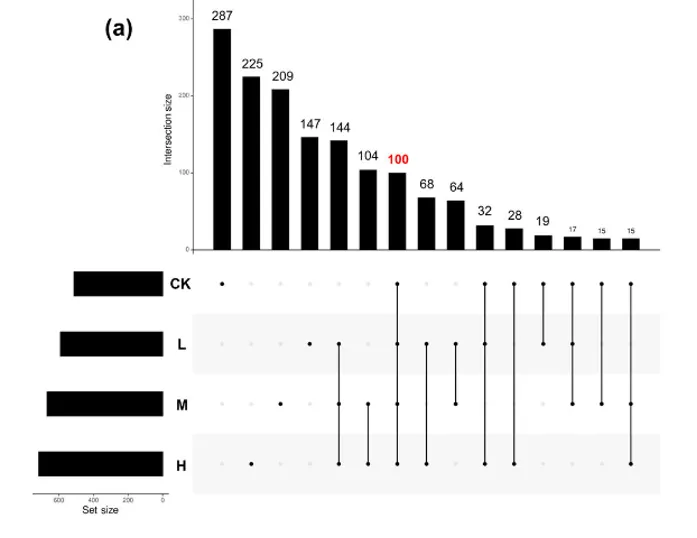
fig.4左下角的长度代表每个As污染水平下的总扩增子序列变异数( ASV )。黑色矩形在水平坐标轴上方的高度代表了对应分组中共同或独特的ASV数。每个黑色矩形下的黑色圆圈将对应的ASV分类为不同的As浓度。
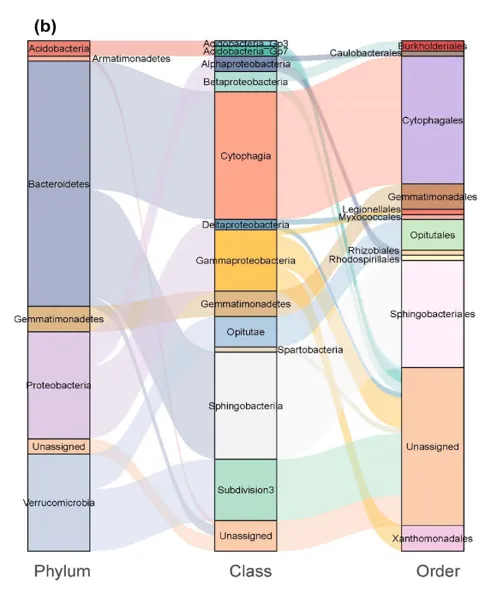
( b )核心微生物组在门、纲、目水平上的相对组成。线条表示之间的连接关系
不同砷污染水平下,菌丝际筛选出的ASV总数随污染变化,对照最少,高砷组最高,且 有100 个 ASV 在 “对照、低、中、高砷” 所有条件下,都能在菌丝际显著富集,说明这些 ASV 不依赖某一种砷浓度,而是稳定存在于菌丝际,因此是 “核心微生物组” 的核心组成,主要属于拟杆菌门、变形菌门和疣微菌门
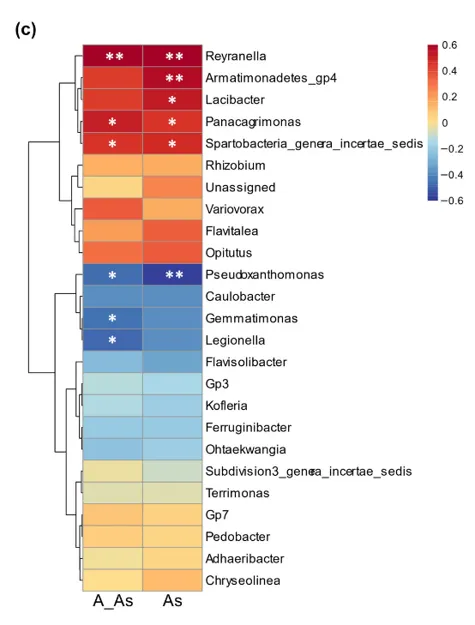
(c )热图显示了土壤中总砷( T-As )和有效砷( A-As )浓度与核心微生物组属丰度之间的相关性。统计学意义( *,P < 0 . 05 )。* *,P < 0 . 01 )采用Spearman相关分析
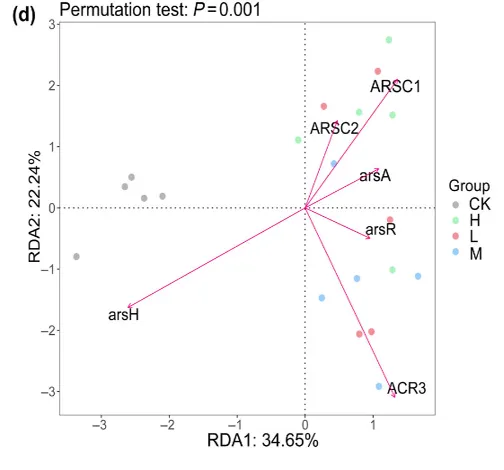
( d ) As生物转化相关的6个功能基因与不同As浓度下菌丝际细菌群落的冗余分析( Redundancy analysis,RDA )
再通过分析核心微生物组成员与砷的关系,4 个核心细菌属的数量,既和总砷相关,也和有效砷相关(如假单细胞属,均来自于变形菌门或拟杆菌门);17 个属和砷含量无显著关联(维持基本功能)。
核心微生物组携带 6 种砷转化相关基因(如 arsC、arsH(亚硝酸盐氧化酶)、ACR3);这些基因代表了砷转化的多种潜在途径,涉及还原、氧化、外排和细胞内解毒。对照土与污染土的核心菌群沿轴 1 明显分开,说明污染显著改变了核心菌群的功能;中污染土与 ACR3 (砷外排)基因关联最强,高污染土与 arsC (还原酶)基因关联最强。代表核心菌群随污染强度调整功能基因表达
结合连续提取(BCR)定量了土壤中的砷组分。
与非菌根土壤相比,菌根土壤中可交换/酸溶性砷(F1)和可还原砷(F2)的比例始终较低,同时可氧化砷(F3)相对富集。这种组分变化符合上述预测功能:在中、高组核心菌群(变形菌门为主)的arsC/ACR3基因潜力强,表明菌群通过表达这些基因,将酸溶态五价砷(As⁵⁺)还原为三价砷(As³⁺),再通过ACR3基因外排到细胞外,或与土壤颗粒结合 ,最终体现为 BCR 分级中酸溶态砷减少,还原态 / 残渣态砷增加。
综合来看,这些结果表明菌根核心微生物群通过在不同地球化学组分间重新分配砷,而非改变总浓度来适应砷污染。这些结果证实了本文的第二个假设,证明了一组核心菌群在砷梯度中持续存在,并可能在菌根环境中对微生物砷生物转化发挥稳定作用。
5.AM 真菌与菌丝际微生物组的共现网络分析
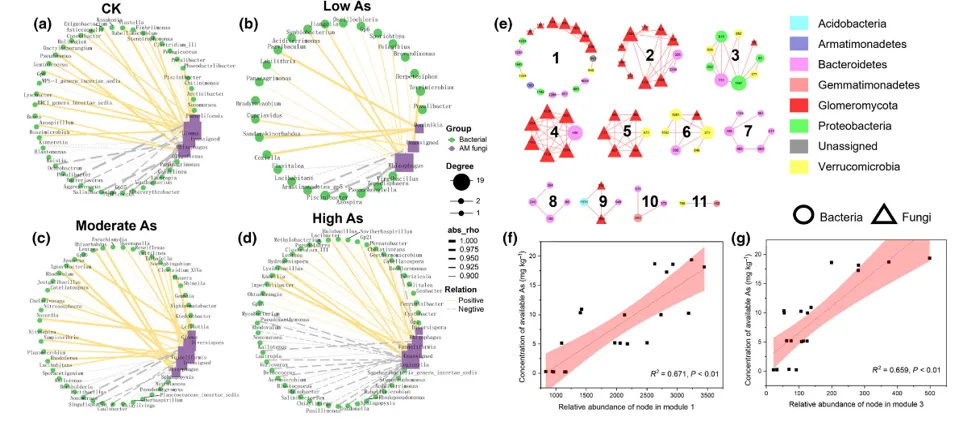
fig.5
(a-d )不同As浓度下菌丝际细菌群落与AM真菌在属水平上的共现网络。节点代表单个菌属,边代表微生物菌属之间的相互作用。
( e )菌丝际核心微生物组与AM真菌的共现网络。基于网络模块对网络进行可视化,数字代表11个模块。圆形节点代表核心微生物组扩增子序列变异体( ASVs ),三角形节点代表AM真菌的ASVs,节点上的编号对应ASV标识符。红色边表示正交互作用,蓝色边表示负交互作用。 不同的颜色代表不同的门。
( f )模型1与土壤有效态As浓度之间的线性拟合。
( g )模块3与土壤有效态As浓度之间的线性拟合。
阴影部分表示拟合的线性回归模型的95 %置信区间(结果可靠性范围)
(a-d)图,随着砷污染浓度的增强,线条连接数与节点数均增多,同时负相关比例上升,但仍是正相关主导,说明从最低浓度时的AM真菌与菌丝际细菌适配性低到中高浓度时微生物适应胁迫并形成协作,互动变得更为频繁。
在e图中,有七个模块同时包含 AM 真菌和核心细菌,且图中几乎全是红线条,说明正相关占主导地位,说明核心微生物组与AM真菌之间以合作为主要互动方式。
同时发现在模块1与模块3中起主导作用的均为携带砷转化基因的变形菌门。
结合统计学线性回归模型验证,它们与有效砷浓度的关联满足 “强相关 + 高显著”:R²≈0.66(接近 1,关联紧密)、P<0.01(结果不是偶然)。意味着 “砷污染越重,它们越活跃,契合 “应对胁迫” 的功能定位,证明其活跃度和砷转化需求直接挂钩。
模块1和模块3可能在菌根真菌与核心菌根圈微生物的协同作用下,对砷的生物转化起关键作用。这两个模块均由变形菌门(Proteobacteria)构成,该门类已知携带砷酸盐还原(arsC)、亚砷酸盐氧化(arsH)及砷外排(arsB/ACR3)相关基因,这些基因可改变砷的溶解度与毒性。
基于共现模式、功能基因预测以及ASV丰度相关性等验证,以及根据之前的研究提出菌根真菌(AMF)菌丝通过分泌物(如糖类和有机酸)富集菌丝体微环境,从而促进砷转化变形菌的生长和代谢活性;反过来,这些细菌可能通过还原、氧化或外排过程缓解局部砷毒性,进而支持菌根真菌在胁迫条件下的进一步定植和生长。
综上所述,这些结果为本文的第三个假说提供了直接证据,即菌根真菌与菌丝体微生物组通过特定的核心分类群模块协同作用,介导砷胁迫下微生物响应和生物地球化学功能的调控


 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
