中国农业大学新污染物团队CEJ:生物炭驱动钴铁氧体中Fe(II)/Fe(III)和Co(II)/Co(III)循环强化类芬顿催化降解抗生素
- 2026-05-28 07:15:06




第一作者:甘睿
通讯作者:李思
通讯单位:中国农业大学

图片摘要
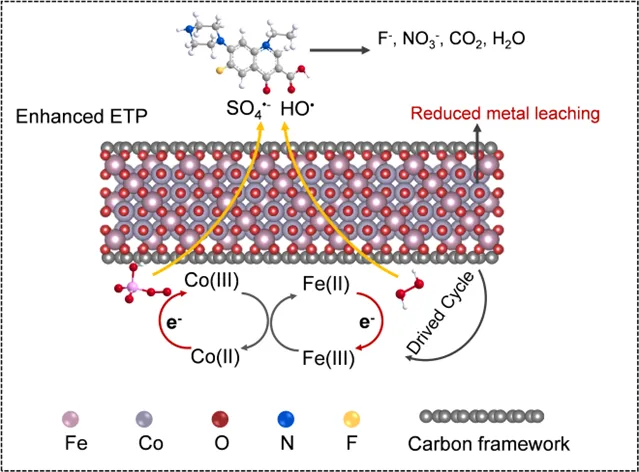

成果简介
近日,中国农业大学资源与环境学院李思副教授课题组在Chemical Engineering Journal上发表题为“Biochar-driven cycling of Fe(II)/Fe(III) and Co(II)/Co(III) in cobalt ferrite for efficient norfloxacin degradation via Fenton-like pathway”的论文。本研究成功合成了一种生物炭负载钴铁氧体(CoFe2O4-BC)复合催化剂,揭示了生物炭作为“电子穿梭体”加速Fe(II)/Fe(III)和Co(II)/Co(III)氧化还原循环的关键机制,实现了中性pH条件下对诺氟沙星的高效降解。该研究为抗生素污染治理提供了新思路。
引言
抗生素污染已成为全球性环境问题,传统水处理工艺对诺氟沙星(NOR)等抗生素去除效果有限,因此亟需开发高效、稳定的深度处理技术。高级氧化技术(AOPs)凭借产生强氧化性活性氧物种的能力,在降解有机污染物方面展现出显著优势。其中,非均相催化剂活化过硫酸盐或过氧化氢的方法因能耗低、可回收等优势受到广泛关注。
钴铁氧体(CoFe2O4)作为一种磁性尖晶石材料,兼具钴离子的高氧化还原电位和铁离子的过氧化氢活化能力,但存在易团聚、电子传递效率不足等问题。生物炭(BC)因其比表面积大、官能团丰富以及成本效益高,常用作载体提高金属纳米催化剂的分散性并减少金属浸出。此外,BC表面的羰基等官能团可作为电子接受位点,参与过硫酸盐活化和活性物种生成。尽管BC被广泛用作催化剂载体,但其调控过渡金属中心氧化还原循环的机制仍未得到充分理解。
基于此,本研究合成了玉米秸秆衍生的CoFe2O4-BC复合材料,系统对比了其活化过氧化氢(H2O2)和过一硫酸盐(PMS)降解NOR的效能。通过实验分析和表征,揭示了BC促进Fe(II)/Fe(III)和Co(II)/Co(III)循环的机理。基于液相色谱-质谱联用(HPLC-MS)技术分析了NOR的降解途径,并通过监测F⁻和NO3⁻的释放来追踪氟和氮的转化。最后,在天然和模拟废水环境中验证了催化体系稳定性。

图文导读
催化剂表征
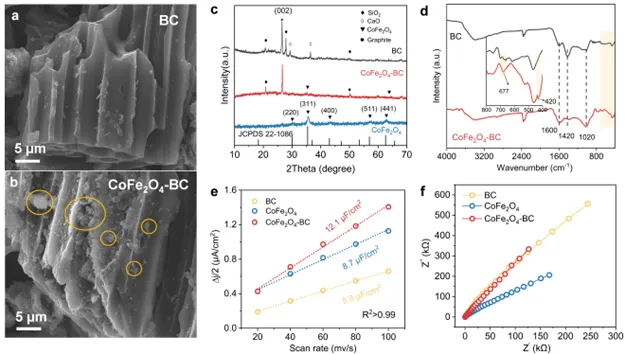
图 1. BC、CoFe2O4和CoFe2O4-BC的SEM图像(a和b)、XRD(c)、FTIR(d)、电化学双层容量(e)和EIS曲线(f)
基于循环伏安法(CV)测量得到的双电层电容(Cdl)估算了材料的电化学活性表面积(ECSA)。如图1e所示,CoFe2O4-BC复合材料的Cdl值最高,为12.1 μF/cm2,是纯CoFe2O4(8.7 μF/cm2)和原始BC(5.8 μF/cm2)的1.4倍和2.1倍,表明其具有更高的活性位点密度。电化学阻抗谱(EIS)证实了CoFe2O4-BC具有更低的阻抗以及更高的传质能力,为类芬顿反应的高效进行奠定基础。
类芬顿催化性能
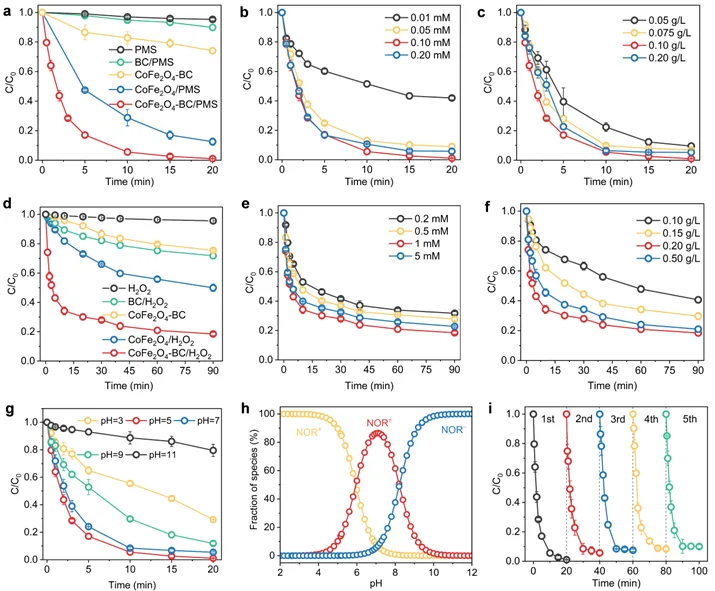
图2. PMS体系对NOR的去除效果(a)、PMS浓度的影响(b)、催化剂用量的影响(c); H2O2体系对NOR的去除效果(d)、H2O2浓度的影响(e)及催化剂用量的影响(f);初始pH值对NOR去除的影响(g)、不同pH值下NOR解离物种的形态分布(h),催化剂的循环性能(i)
在PMS活化体系中,CoFe2O4-BC在20分钟内可以几乎完全去除NOR,反应速率常数k为0.289 min-1,分别是CoFe2O4和原始生物炭的2.78倍和47.7倍。弱酸性至中性(pH 5~7)催化性能最优,pH降至3时k值降至0.058 min-1。pH升至9或11时k值分别降至0.105和0.010 min-1,归因于静电排斥、SO4•⁻转化为•OH(氧化电位降低)及钴氢氧化物沉淀钝化表面。CoFe2O4-BC/ H2O2的k值较CoFe2O4和BC分别提高4.8倍和8.5倍。但整体降解效率低于PMS体系,且对pH依赖不同(pH=3最优)。
类芬顿催化机理研究
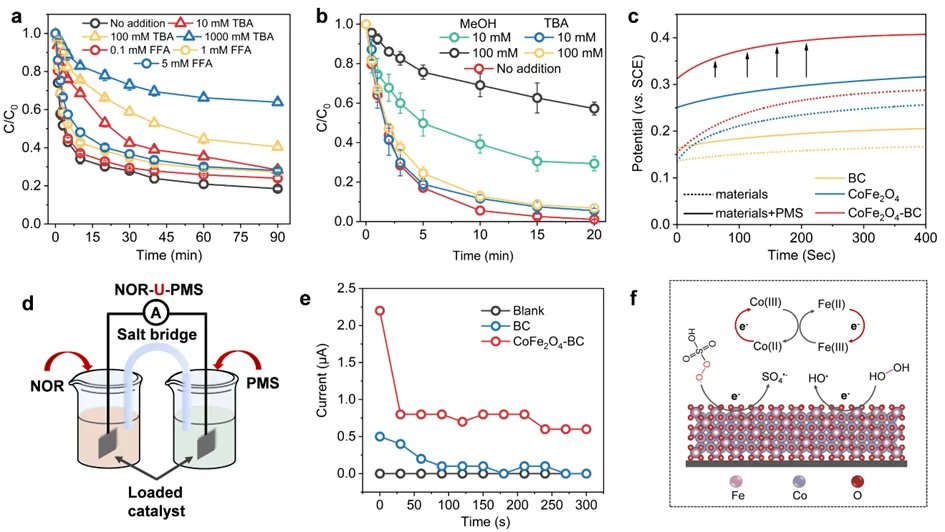
图3. CoFe2O4-BC/H2O2(a)和CoFe2O4-BC /PMS (b) 体系中活性氧清除剂对去NOR去除的影响,OCP (c)、盐桥实验 (d)、双室电化学系统 i-t 曲线(e), CoFe2O4-BC 活化 PMS/H2O2的机理示意图(f)。
在CoFe2O4-BC/PMS体系中,SO4•⁻对NOR降解的贡献为50.9%,非自由基路径为43.3%,•OH仅贡献5.8%。在H2O2体系中,•OH贡献26.9%,非自由基路径占73.1%。盐桥实验的结果表明,将NOR和PMS分置两个反应池,CoFe2O4-BC的电流响应显著高于BC,证明复合材料促进了电子转移过程。
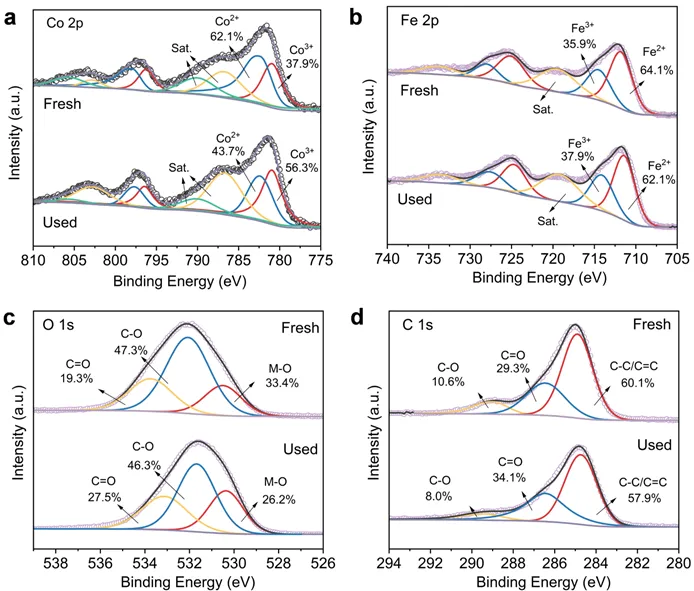
图4.反应前后CoFe2O4-BC 的 Co 2p(a)、Fe 2p(b)、O 1s(c)和 C 1s(d)的 XPS 光谱。
通过XPS分析了反应前后CoFe2O4-BC表面化学态变化。Co(II)含量从62.1%降至43.7%,Co(III)从37.9%升至56.3%,说明Co(II)是PMS活化的主要电子供体。Fe(II)含量从64.1%降至62.1%,Fe(III)从35.9%升至37.9%,变化幅度小于Co,表明Fe主要起协同作用,通过促进Co(III)/Co(II)的循环再生Co(II)。反应后C 1s和O 1s谱图中,C=O比例增加,C-O和M-O比例减少,表明生物炭表面含氧官能团作为电子供体,协助还原Co(III)/Fe(III),维持体系中氧化还原循环。综上所述,BC不仅分散金属纳米颗粒,更直接参与电子传递,从而大幅提升催化效率。
NOR降解途径和毒性分析
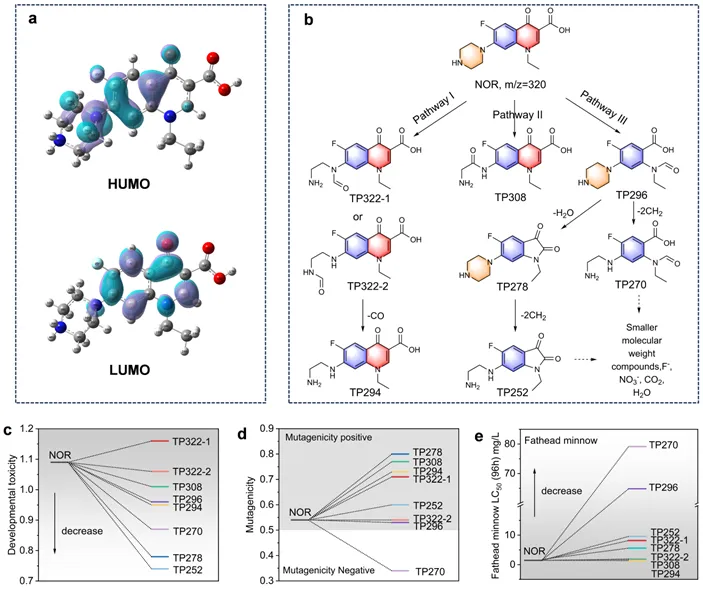
图 5.NOR的HOMO和LUMO(a),NOR的降解途径(b),NOR及其降解产物的发育毒性(c)、致突变性(d)和黑头鲦鱼毒性(e)。
NOR分子的最高占位分子轨道(HOMO)主要位于哌嗪环和邻近的苯环(C4-N26)上,表明这些区域易受亲电自由基(SO4•⁻、•OH)攻击。Fukui指数显示哌嗪环上N26亲电反应活性最高。基于HPLC-MS鉴定了NOR的降解产物,NOR通过哌嗪环羰基化生成TP322,并进一步脱羰基生成TP294。此外,NOR还可通过喹诺酮环氧化脱羧生成TP296,随后通过开环反应产生TP270、TP252等。部分产物经进一步氧化最终矿化为CO2、H2O,同时释放F⁻和NO3⁻。值得注意的是,降解过程中可能生成毒性更高的中间体,因此实际应用需保证足够长的反应时间以实现深度矿化,而非仅关注母体污染物去除。
实际应用和环境影响评估
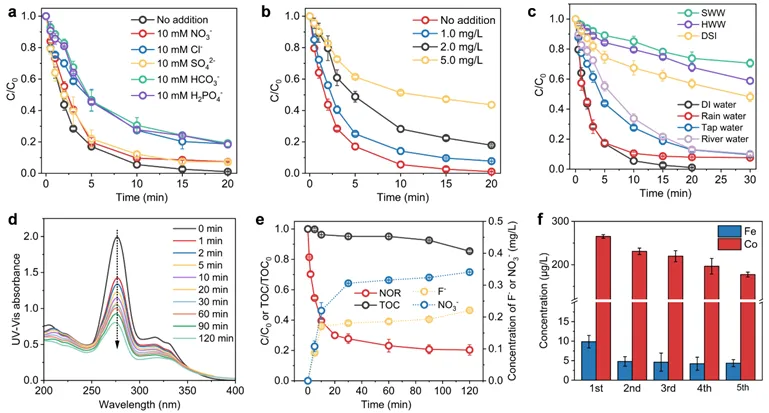
图6.无机阴离子(a)、HA(b)、天然和模拟废水环境(c)中体系的催化性能,紫外可见吸收光谱(d)、矿化率以及F⁻和NO3⁻的转化(e)和金属浸出(f)。
CoFe2O4-BC/PMS体系在雨水中降解NOR的效果与去离子水相当,在自来水和河水略有下降。模拟养猪废水、医院废水、生活污水中的抑制较为明显(去除率分别下降约30%~50%),主要由于高浓度阴离子、高pH值及有机物(尿素、葡萄糖等)竞争。催化剂中钴(<0.2 mg/L)和铁(<0.01 mg/L)的浸出量远低于《中国国家地表水环境质量标准》(GB-3838-2002,钴<1.0 mg/L,铁<0.3 mg/L)的允许范围。这一结果表明,CoFe2O4-BC复合材料造成二次金属污染的环境风险较低。氟和氮的化学计量转换率分别为18.6%和2.93%。

小结
本研究成功合成了CoFe2O4-BC复合材料,用于活化H2O2和PMS降解NOR。多孔生物炭载体提供了均匀的CoFe2O4纳米粒子分布,更重要的是,生物炭作为电子穿梭体,加速了Fe(II)/Fe(III)和Co(II)/Co(III)氧化还原循环,显著提高了催化活性和稳定性。NOR的降解主要通过哌嗪环和喹诺酮基团的转化进行。然而,某些降解中间产物的毒性可能超过NOR的毒性。在处理实际废水时,有必要延长反应时间和优化试剂用量,以实现高矿化度和有效解毒。总之,这项工作为抗生素治理提供了可行的思路,并阐明了生物炭在非均相类芬顿催化中调控金属氧化还原循环的关键作用。

作者简介
李思:中国农业大学资源与环境学院副教授,博士生导师。研究领域包括新污染物环境行为、水体和土壤污染控制技术开发与应用等。主持国家自然科学基金等项目。在Water Research,Chemical Engineering Journal, Journal of Hazardous Materials等期刊发表论文50余篇,其中高被引论文6篇。论文被引4500余次,h指数35(Google Scholar)。
第一作者:甘睿,中国农业大学资源与环境学院2025级博士研究生。研究方向为环境功能材料、高级氧化技术。以第一/共一作者在Applied Catalysis B: Environment and Energy, Chemical Engineering Journal, Journal of Hazardous Materials等期刊发表5篇学术论文。
文章链接:https://doi.org/10.1016/j.cej.2026.176157

投稿、合作、转载、进群,请添加小编微信Environmentor2020!环境人Environmentor是环境领域最大的学术公号,拥有25W+活跃读者。由于微信修改了推送规则,请大家将环境人Environmentor加为星标,或每次看完后点击页面下端的“在看”,这样可以第一时间收到我们每日的推文!环境人Environmentor现有综合群、期刊投稿群、基金申请群、留学申请群、各研究领域群等共20余个,欢迎大家加小编微信Environmentor2020,我们会尽快拉您进入对应的群。

往期推荐



扫描二维码,快速入群~
随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 中国农业科学院国家南繁研究院招聘公告
- 印度农业科技公司 PI 启用新 LOGO
- 吉林农业大学校企合作平台——“北方重要食药用菌种质资源开发与利用联合中心”年度工作总结会日前举行
- 《中医农业五位一体技术研发中心赴济南市鸿德中医院考察调研》
- 本科毕业于东北农业大学,博士毕业于哈尔滨工业大学,东北农大副教授以第一作者身份在一区Top期刊上发表论文
- 青岛农业大学青年教师一作发表Nature Genetics研究论文
- 华南农业大学夏瑞课题组揭示LcTINY-LcbZIP53模块调控荔枝种子大小的分子机制
- 从海绵城市到海绵农业
- 警惕!农业诈骗套路!
- 优质工薪专属!农业银行【随薪贷】最高200万,纯信用无抵押,收入稳定即可申!
