背景介绍
从废塑料中回收碳资源与能量,是将闲置资源纳入循环经济的有效途径。化学升级转化可将难降解塑料高值化为高附加值化学品与绿色能源载体,其中无溶剂、无氢气条件下的裂解-催化升级转化,是拆解废塑料制备高附加值产物的创新技术路线。
碳纳米管(CNTs)在电催化、超级电容器等领域应用广泛,具有重要价值。电解水制氢是制备高纯H2的理想方式,但碱性电解液中阴极析氢反应(HER)动力学缓慢,急需高效、稳定、低成本的催化剂。镍基电催化剂因价态可调、本征活性高且可发生自氧化,在碱性HER中优势显著,但其在氧化、腐蚀环境下难以维持活性与结构稳定性。Ni包裹CNTs电催化剂凭借碳壳与金属核的强界面耦合作用,可促进电荷转移、保护活性位点,但由于制备工艺复杂,活性组分随机分布,限制了其规模化应用。
本文提出可控、无溶剂、无H2解聚策略(CSHFD),利用有序微孔碳负载镍基纳米颗粒(Ni-OMC),实现废塑料选择性转化为具有分级孔道结构的功能化纳米复合材料(Ni-CNTs-OMC)直接用作HER电催化剂,既联产了高纯度绿色H2,又搭建起热催化与电催化的桥梁。实验表明Ni20-OMC在纳米复合材料生成和H2产生方面表现出优异的双功能催化活性,实现55.26 mmol g-1 plastic的H2产率,H2占比达86.86 vol%。Ni20-CNTs-OMC在10 mA∙cm-2电流密度下过电位仅215 mV,塔菲尔斜率109 mV∙dec-1,经15000次循环后仍保持稳定。Ni包裹CNTs结构通过优化电子分布、降低水解离能垒、调控*H吸附提升HER性能。综上,本工作提供了可控可持续的路径,可将实际废塑料转化为高性能电催化剂与高纯度H2。
图文导读
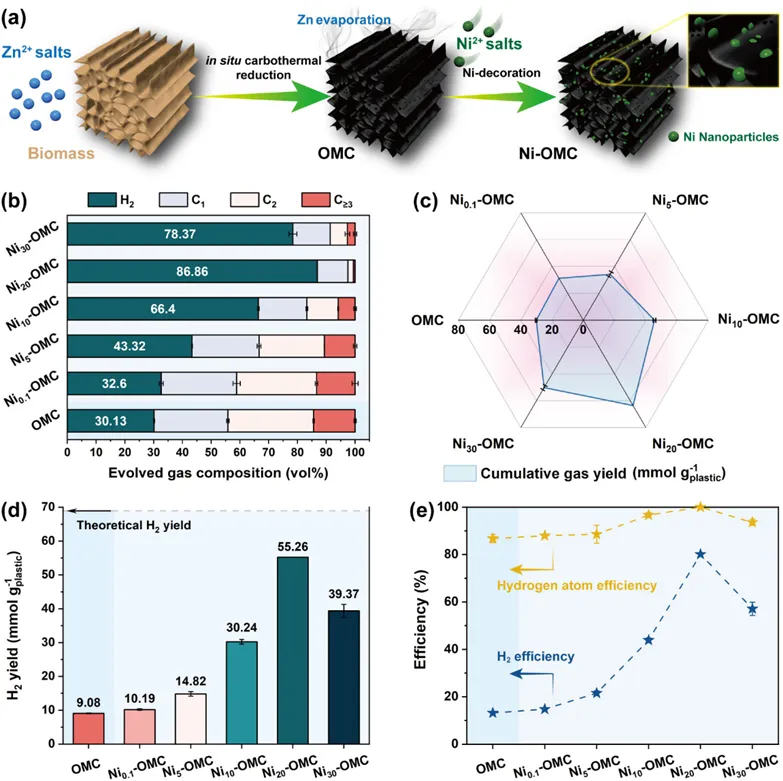
Figure 1. Gas formation behaviors derived from CSHFD of PE over Ni-OMC: (a) schematic diagram of the synthesis of Ni-OMC; (b) evolved gas compositions; (c) cumulative gas yield; (d) H2 yield; and (e) H2efficiency and hydrogen atom efficiency.
如图1a所示,Ni-OMC催化剂通过原位碳热还原结合Ni负载法制备而成。Ni-OMC催化剂上的产氢量随Ni负载量的增加呈现火山型变化趋势。更多Ni NPs负载到微孔碳骨架上时,H2浓度显著升高,这与C1、C2及C≥3轻质烃的浓度变化相反(图1b),表明产氢量的提升以牺牲轻质烃气体为代价。碳骨架上更多的Ni NPs活性位点具有更强的C–C键与C–H键断裂能力。因此,随着Ni-OMC中Ni NPs负载量的增加,累积气体产率与H2产率均显著提升(图1c-d)。当Ni负载量过高(30 wt%)时,活性Ni NPs发生团聚,产氢量不再进一步提升。当Ni负载量为20 wt%时(Ni20-OMC)达到最高富氢气体产率(63.62 mmol g-1 plastic),较纯OMC(30.13 mmol g-1 plastic)提升了1.1倍以上(图1c)。此外,Ni20-OMC的产氢量为55.26 mmol g-1plastic,氢气体积占比为86.86 vol%;其他Ni-OMC催化剂的产氢量为9.08~39.37 mmol g-1plastic,氢气体积占比为30.13~78.37 vol%(图1b、d)。Ni20-OMC表现出极高的H2产率与氢原子效率,分别达到80.14%和100%(图1e)。上述结果共同表明原始PE中的氢元素已完全转化为H2及其他气体,而大部分碳元素则在催化剂表面转化为固相碳材料。
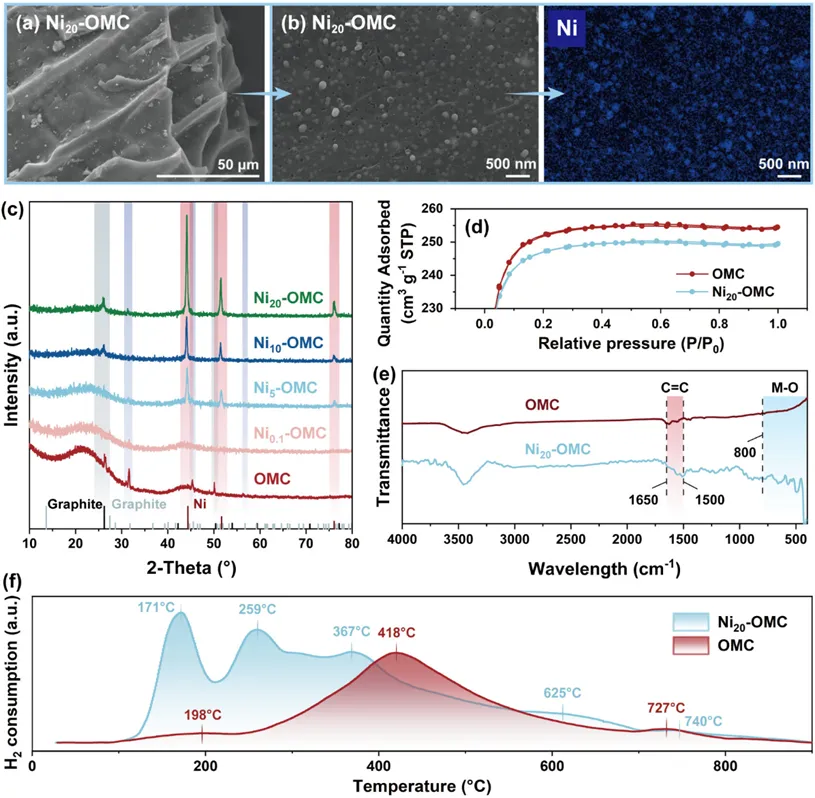
Figure 2. Physicochemical characteristics of Ni-OMC: (a-b) SEM and EDS images; (c) XRD patterns; (d) N2adsorption–desorption isotherms; (e) FTIR spectra; and (f) H2-TPR profiles.
如图2a-b所示,Ni20‑OMC呈现出均匀的纤维束状结构,与未负载Ni的前驱体OMC形貌相似。其表面富含由ZnCl2作用形成的微孔结构。EDS分析表明Ni NPs在Ni20‑OMC催化剂表面均匀分布与富集且Ni NPs平均粒径小于30 nm,均匀嵌入微孔碳骨架中,有利于提升活性位点分散度、提高反应效率并降低催化剂失活风险。XRD图谱(图2c)中在44.3°、51.7°和76.1°处的衍射峰,分别对应金属Ni0的(111)、(200)和(220)晶面(JCPDS 89‑7128),表明原位碳热还原过程提供了还原氛围,确保NiO充分转化为金属态Ni0。将Ni NPs含量提高至20 wt%时,由于过量Ni物质的分散与侵入堵塞部分微孔,使得BET比表面积略有下降(图2d)。如图2e所示,Ni20‑OMC中C=C红外峰强度显著高于纯OMC,表明其石墨化程度更高。此外,Ni20‑OMC在400-800 cm-1区间内对应金属氧化物的峰更强,说明体系中存在大量Ni物质。H2‑TPR曲线结果显示(图2f),与纯OMC相比,Ni20‑OMC还原温度更低,表明其还原性更强,说明Ni20‑OMC在反应初期可提供更多活性位点。
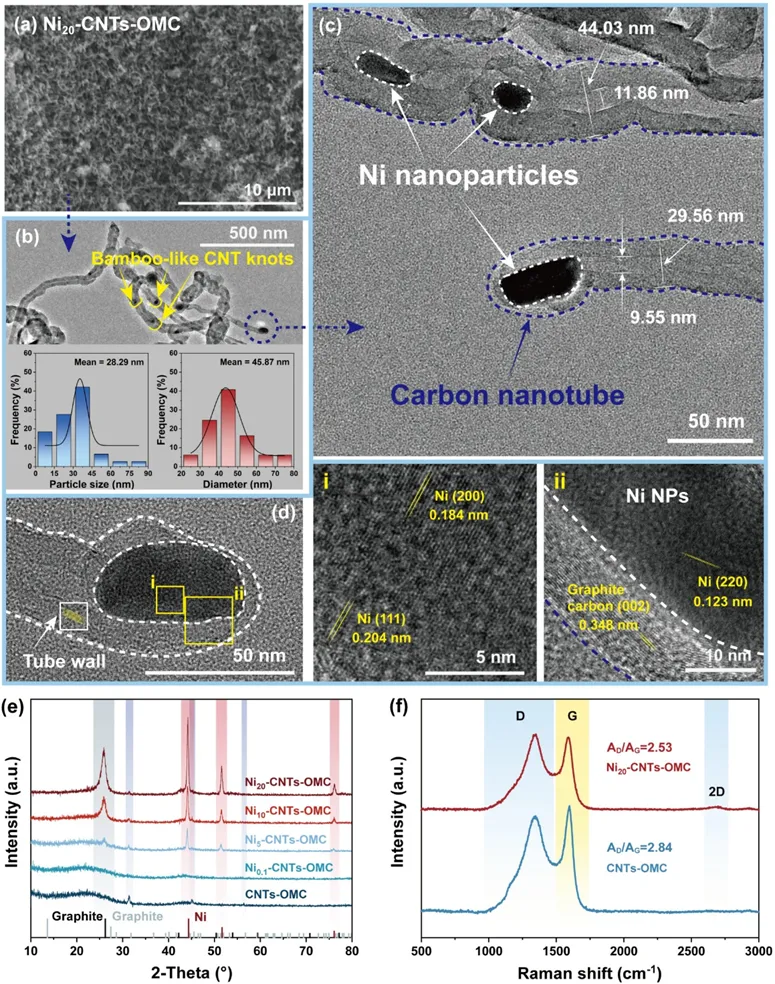
Figure 3. Characterization of functionalized Ni-CNTs-OMC: (a) SEM image; (b-d) TEM images; (e) XRD patterns; and (f) Raman spectra.
生长于Ni-OMC表面碳产物的表征结果显示,随着Ni NPs含量增加,催化剂表面观察到扭曲缠绕的丝状碳明显增多(图3a),其中Ni20‑OMC表面的碳沉积量最为丰富。由TEM(图3b)可知生长于Ni20‑OMC表面的丝状碳呈现管壁光滑、中空结构,并伴有少量竹节状节点,其被确认为CNTs,平均直径为45.87 nm(图3b-c)。高分辨TEM图像(图3d)显示,所锚定的CNTs由多层石墨层构成,晶面间距为0.348 nm,对应石墨的(002)晶面。晶面间距0.123、0.184和0.204 nm分别对应金属Ni NPs的(220)、(200)和(111)晶面。此外,金属Ni NPs位于多壁CNTs的顶端或管芯内部(图3b-d),表明其主要为顶端生长。由上述结果可知PE链首先吸附在布朗斯特酸位点上,形成C=O中间体,随后通过可控热裂解发生C–C键断裂,生成短链自由基。在催化重整阶段,短链烃在金属Ni活性位点上进一步发生C–C键与C–H键断裂。生成的碳原子在金属Ni NPs内部扩散,最终在Ni NPs底部成核并析出,形成CNTs。剩余的氢原子重新结合,以H2的形式脱附。在XRD图谱(图3e)中约26.2°处出现对应石墨相的衍射峰,证实催化剂上形成了石墨化CNTs。Ni20‑CNTs‑OMC的峰强度最高,表明其石墨化碳生成量最多。拉曼光谱(图3f)显示在1340、1590和2700 cm-1附近检测到分别对应D峰、G峰与2D峰的特征峰。催化剂表面生长的固相碳均呈现出明显的G峰与2D峰,证实CNTs的形成。D峰强度较高表明CNTs内部包裹了金属Ni NPs。此外,Ni20‑CNTs‑OMC的AD/AG峰面积比高达2.53,进一步证实CNTs内部包裹了大量金属Ni NPs。相较下,Ni20‑CNTs‑OMC的AD/AG比值相对更低,表明其表面负载的CNTs具有更高的石墨化程度。综上,Ni20‑OMC催化剂在制备高纯H2与功能化Ni‑CNTs‑OMC纳米复合材料方面表现出优异的催化性能,是一种极具应用前景的催化剂。
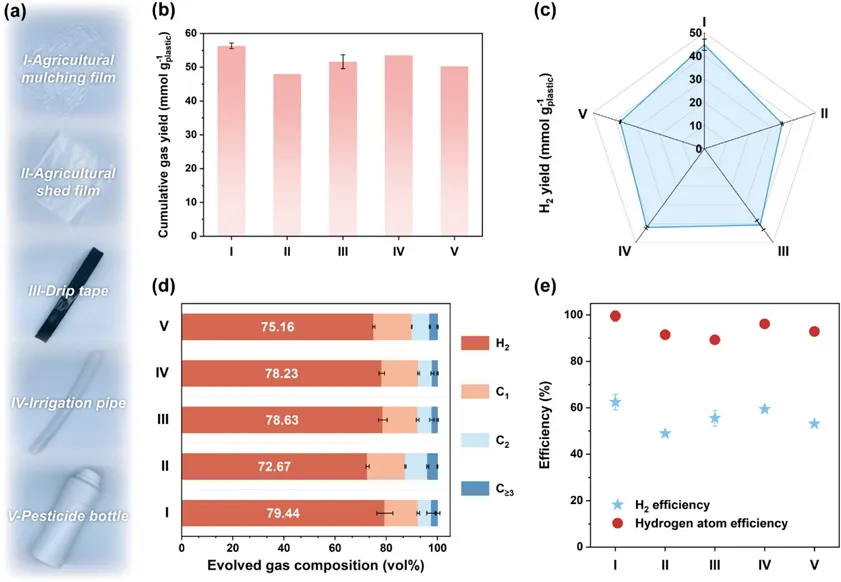
Figure 4. Upcycling of real-life PE waste through the CSHFD strategy: (a) photographs of agricultural mulching film (I), agricultural shed film (II), drip tape (III), irrigation pipe (IV), and pesticide bottle (V); (b) cumulative gas yield; (c) H2 yield; (d) evolved gas compositions; (e) H2 efficiency and hydrogen atom efficiency.
采用优化后的Ni20‑OMC催化剂,对实际废弃塑料(图4a)测试可知,由于回收PE中含有各类添加剂(图4b-e),实际废弃PE的累积产气量和产氢量均低于纯PE。在各类实际废弃PE中,添加剂含量较少的PE‑I表现出更高的累积产气量(56.36 mmol g-1 plastic),H2体积占比可达79.44 vol%(图4b、d)。此外,经CSHFD工艺处理的PE‑I具有较高的H2产率(62.42%)和氢原子效率(99.53%),表明PE‑I中的氢几乎被完全提取并转化为高纯度H2与轻质烃(图4e)。上述结果表明,由于双反应器设计可分离难熔灰分并减缓催化剂失活,使得Ni20‑OMC催化剂能够高效转化实际废弃塑料,并对添加剂和杂质具有良好耐受性。
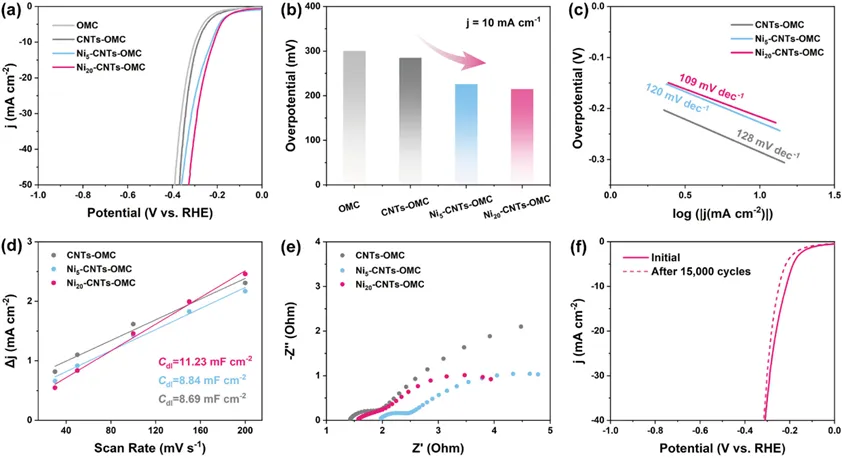
Figure 5. Electrochemical performance of Ni-CNTs-OMC nanocomposites in the alkaline HER: (a) LSV curves with 90% iR correction; (b) overpotentials at 10 mA cm–2; (c) Tafel plots; (d) Cdl; (e) Nyquist plots; and (f) 15,000-cycle stability test of Ni20-CNTs-OMC.
采用标准三电极体系,在1.0 M KOH电解液中,以Ni负载量为变量,评估Ni-CNTs-OMC的碱性HER活性。经iR校正的LSV曲线(图5a)表明,Ni20-CNTs-OMC表现出最优的电催化活性,在电流密度达到10 mA cm-2时,η10仅为215 mV,显著优于OMC(η10=300 mV)、未负载Ni NPs的CNTs-OMC(η10=285 mV)以及Ni5-CNTs-OMC(η10=226 mV)(图5b)。Tafel斜率分析结果显示Ni20-CNTs-OMC的塔菲尔斜率为109 mV dec-1,小于CNTs-OMC(128 mV dec-1)和Ni5-CNTs-OMC(120 mV dec-1),表明该催化剂可促进电荷转移与质量扩散(图5c)。上述结果表明催化剂上的析氢反应主要遵循Volmer–Heyrovsky机理,且初始放电过程为速率控制步骤。通过在非法拉第电流区、不同扫描速率下进行循环伏安(CV)测试(图5d),证实Ni20‑CNTs‑OMC具有较高的双层电容值(Cdl=11.23 mF cm-2),表明其表面可接触的活性中心数量更多。EIS(图5e)分析显示Ni20‑CNTs‑OMC的电荷转移电阻(Rct)显著更低,进一步证明包裹金属Ni NPs、高石墨化程度的CNTs结构,能够有效促进界面电子转移并提升导电性。在1.0 M KOH电解液中经过15000次循环伏安(CV)测试后,Ni20‑CNTs‑OMC的LSV曲线仅表现出可忽略的过电位偏移(图5f),表明该结构具有优异的稳定性,在多壁碳纳米管长期碱性HER过程中,可有效保护金属Ni NPs活性中心的结构与催化完整性。
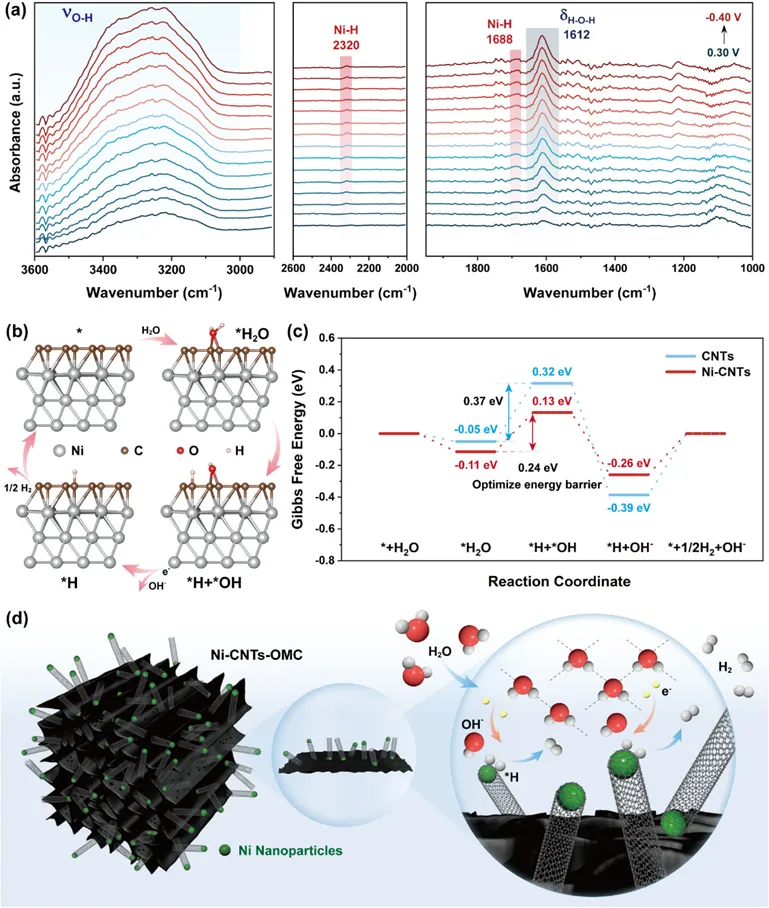
Figure 6. In situ characterization and DFT calculations of the Ni-CNTs-OMC nanocomposite electrocatalyst in the alkaline HER: (a) in situ ATR-SEIRAS spectra of interfacial H2O and hydrogen intermediate (*H) on Ni20-CNTs-OMC; (b) HER mechanism on Ni-CNTs; (c) Gibbs free energy for the HER on CNTs and Ni-CNTs; and (d) schematic illustration of the proposed HER enhancement mechanism on Ni-CNTs-OMC.
在in situ ATR-SEIRAS光谱中(图6a),1612 cm-1处出现特征H–O–H弯曲振动(δH–O–H),在3000-3600 cm-1处出现O–H伸缩振动(νO–H)。此外,随着施加电位向负方向移动,δH–O–H峰出现5 cm-1蓝移,表明界面氢键网络增强,从而加速质子转移与HER反应动力学。νO–H伸缩振动区域显示吸附在Ni20‑CNTs‑OMC表面的水分子发生6 cm-1红移,说明O–H键被削弱、水的解离过程得到促进,催化剂表面加速了速率决定步骤,从而加快了整体HER反应动力学。在0.3至-0.4 VRHE的电位区间内,Ni20‑CNTs‑OMC在约3300 cm-1处出现显著吸收峰,反映出电化学双电层内氢键网络得到增强。在0.2 VRHE处出现2320 cm-1的特征Ni–H伸缩振动峰,说明Ni20‑CNTs‑OMC中Ni与*H中间体之间的强相互作用有利于氢的吸附与后续还原过程。随着电位负移,Ni–H峰强度逐渐增强,表明*H中间体的吸附量不断增加。在阴极极化条件下,约1688 cm-1处的另一Ni–H振动峰也逐渐增强,表明多个金属中心被有效活化,进一步加快HER反应动力学。基于H2O在碳表面解离的优化电催化剂模型(图6b),得到其吸附自由能(图6c)结果显示Ni-CNTs的H2O吸附自由能(=0.11 eV)优于CNTs(=-0.05 eV),表明Ni-CNTs对H2O具有更强的结合能力,提升了反应物供给效率。此外,H2O在CNTs表面的解离能垒为0.37 eV。相比之下,Ni-CNTs界面促进了Volmer步骤,能垒降至0.24 eV,反应为微弱吸热过程(0.13 eV),表明Ni封装碳纳米管结构可在Ni-CNTs界面实现强电子相互作用。当Ni纳米颗粒被封装在石墨化碳纳米管内部后,Ni-CNTs表现出接近最优的热力学行为(ΔG*H=0.26 eV),相比之下,纯CNTs对氢的吸附更强(ΔG*H=0.39 eV)。由上述结果可知富电子的Ni-CNTs界面位点成为催化活性中心,在HER过程中促进电子从电催化剂向反应中间体转移(图6d)。







 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
