Highly efficient production of 1,5-pentanediol from furfural hydrogenation over Co-Fe/ZrO₂ catalysts
在钴铁/氧化锆催化剂作用下,糠醛加氢高效制备1,5-戊二醇https://doi.org/10.1016/j.apcatb.2025.126247糠醛(FF)作为一种大宗且应用广泛的生物质基平台化合物,通过转化为高附加值的戊二醇,在生物基聚合物的可持续生产中具有重要意义。然而,在多功能金属负载型催化剂上,通过开环反应实现1,5-戊二醇(1,5-PeD)的高选择性合成极具挑战性,其难点在于难以平衡加氢脱氧(HDO)与过度加氢反应。为攻克这一难题,本研究开发了钴铁/氧化锆(Co-Fe/ZrO₂)催化剂,用于糠醛选择性一锅法加氢制备1,5-PeD。在优化条件下(150 °C、4.0 MPa H₂、反应8小时、水/异丙醇混合溶剂(体积比10:90)),20Co-5Fe/ZrO₂催化剂实现了78.9%的1,5-PeD高产率,创文献报道最高水平。钴(Co)可促进氢气活化,而氧化铁(FeOₓ)的掺杂显著提升了催化剂的氧空位(Oᵥ)浓度,进而增强了催化活性与1,5-PeD选择性。以糠醇(FA)为模型底物,结合同位素标记实验的动力学研究表明,水介导的质子化在特定C–O–C键的选择性断裂中起关键作用。研究发现,水通过促进质子转移直接参与C–O–C键的断裂过程,激活了一条低能垒反应路径。在催化循环中,金属/水界面自发生成活性氢物种,间接调控反应机理与动力学。活性氢物种与酸性位点的协同作用,实现了糠醛向1,5-PeD的直接、选择性转化。这些发现凸显了通过金属改性调控催化剂结构,引导生物质平台分子加氢路径向目标高附加值产物转化的巨大潜力。木质纤维素作为自然界中唯一的可再生有机碳资源,通过高效转化为高附加值化学品和液体燃料,对实现全球碳中和目标具有战略意义。在众多生物质基平台化合物中,半纤维素衍生的糠醛(FF)因其成熟的工业生产体系,成为合成可持续燃料与化学品的关键可再生中间体。FF主要通过木质纤维素生物质中C5糖的酸催化转化获得,可通过多样化路径选择性转化为多种高附加值产物:FF的胺化反应可定向合成含氮药物中间体(如糠基胺);氧化转化可生成羧酸及开环酸(如呋喃酸、乙酰丙酸);羰基(C=O)加氢生成糠醇(FA),进一步饱和FA中的呋喃环可制得四氢糠醇(THFA)。这些衍生物因低毒性和高溶解性,广泛应用于电子清洗剂、油墨和农用化学品领域。特别地,FF还可通过呋喃环C-O键断裂加氢裂解生成1,5-戊二醇(1,5-PeD)(方案1)。值得注意的是,1,5-PeD与石油基α,ω-二醇具有结构同源性,可直接替代用于工业聚酯、氨纶和聚氨酯生产,兼具显著的经济与环境优势。因此,开发高效催化剂实现FF及其衍生物选择性加氢制备1,5-PeD具有重大实践意义,可打通可持续生物质利用与大宗化学品制造脱碳化的关键路径。
尽管异相催化剂用于糠醛(FF)选择性加氢的研究已广泛开展,但实现1,5-PeD的高选择性合成仍面临重大挑战。当前研究主要聚焦于贵金属催化剂将FF衍生物(FA/THFA)转化为1,5-PeD,而FF直接一步法转化为1,5-PeD的路径尚未充分探索。该转化涉及醛基加氢、呋喃环开环及后续加氢等多步复杂反应,需精确调控反应路径以平衡选择性。传统多步路线从木质纤维素生物质到1,5-PeD需在每步反应后进行高能耗的分离/纯化,因此FF一锅法催化转化为1,5-PeD极具吸引力。然而,该方法需多步序贯反应并抑制竞争路径,严重限制了1,5-PeD的可达产率。作为关键中间体的四氢糠醇(THFA)具有饱和呋喃环结构,其环开裂需高活化能,因此通常需苛刻条件——包括高压氢气(6-8 MPa)、低底物/催化剂比及长反应时间(≥20 h)。从机理上看,环开裂过程由亲氧位点(如MoOx、WOx、ReOx)与加氢活性金属中心(如Rh、Ir、Pt)的协同作用主导。FA的羟基吸附于亲氧位点,邻近金属位点发生H₂异裂生成活性H⁻物种,H⁻对FA的C2位进行区域选择性亲核进攻引发环断裂,随后质子化生成的烷氧中间体生成1,5-PeD。聚焦金属与亲氧位点空间邻近性的催化剂设计策略被证明对优化该路径至关重要。Tomishige等通过FF加氢在Pd(0.66)-Ir-ReOx/SiO₂和Rh(0.66)-Ir-ReOx/SiO₂双金属催化剂上实现了>70%的1,5-PeD产率。例如,Pd-Ir-ReOx/SiO₂采用两阶段工艺:40-60℃下FF首先加氢生成THFA(2 h),随后100-120℃下THFA氢解(24-48 h),最终获得71.4%的1,5-PeD产率。尽管该产率高于一锅法效率,但多步操作的复杂性凸显了开发兼具序贯加氢与氢解活性的创新催化剂架构的必要性。该机制涉及亲氧物种通过羟基功能增强底物吸附,配合邻近Ir-ReOx活性位点协同促进C-O键氢解。尽管选择性显著,但贵金属成本高昂及催化效率有限阻碍了工业化推广。因此,研究转向非贵金属基双功能催化剂,将金属活性中心与富氧空位氧化物整合用于选择性加氢。在镍基催化剂中,Shimazu等[18]开发的Ni-Y₂O₃催化剂在150℃、2 MPa H₂及24 h反应条件下实现42%的1,5-PeD选择性,但THFA共生成显著(30.4%选择性)。Liang等采用Ni/3.0La₂O₃-CeO₂催化剂推进THFA氢解,获得70%的1,5-PeD产率并降低活化能(57.5 kJ/mol)。钴基催化剂在该选择性加氢反应中亦表现活跃。Shao等合成Co-Mg-Al层状双氢氧化物(LDHs)用于FA转化为1,5-PeD(产率35.0%),而Kim等在类似条件下实现47.5%产率。Ding等设计的Ni-Co-Al三金属催化剂实现FA直接转化为1,5-PeD,选择性达42.5%同时生成12.2%的1,2-戊二醇。Xiao等[23]通过碳热冲击(CTS)法成功合成三元金属氧化物催化剂1.5PtCo₁Ce₁-CTS,在170℃、3 MPa H₂反应4 h条件下,该催化剂的1,5-PeD选择性达59.2%。Xiang等报道的Co-CoOₓ催化剂(源自Co-MOF前驱体)在170℃、3 MPa H₂下反应1.5 h,表现出46%的1,5-PeD选择性,同时将1,2-戊二醇的形成抑制至3.74%。Co-CoOₓ的分级花状二维纳米片结构协同结合了Co纳米颗粒与富氧空位CoOₓ,通过MOFs中Co²⁺-配体配位最大化活性位点密度。尽管取得这些进展,非贵金属催化剂在从FF及其衍生物最大化1,5-PeD产率方面仍面临挑战,需进一步优化双功能位点架构与反应动力学,以缩小实验室性能与工业需求间的差距。因此,开发高效且经济的非贵金属催化体系用于FF直接氢解生成1,5-PeD兼具基础研究与工业意义。
水作为环境友好型溶剂,是生物质转化过程中普遍存在的介质,在溶解、传热和传质中发挥关键作用。其存在显著调控催化活性、选择性和稳定性。然而,水功能的原子级机制仅在少数案例中得以阐明,其直接参与催化反应的实验证据仍稀缺。Dumesic等证明Rh-Re/C催化剂表面的铼(Re)原子可活化水分子生成酸性位点,这对C-O键氢解至关重要。Merte等进一步揭示,水形成致密微环境,通过氢溢流在金属活性中心和反应物结合位点间重新分配电荷。值得注意的是,水作为氢供体直接参与关键反应动力学相关步骤的案例已有报道,包括水辅助糠醇开环生成1,2-戊二醇、乙酰丙酸加氢生成γ-戊内酯及糠醛加氢生成糠醇。当前研究主要依赖分子动力学模拟或密度泛函理论计算等间接表征手段探究水的溶剂效应。因此,获取直接光谱证据对机制验证至关重要。
本研究通过共沉淀-水热合成法成功构建了具有协同催化界面的Co-Fe/ZrO₂催化剂。该设计实现了加氢活性位点与酸性位点的纳米尺度空间耦合,显著提升了FF氢解生成1,5-PeD的一锅催化效率。与Co/ZrO₂的系统对比表明,Fe掺入(FeCo=0.25)有效调控了金属钴的电子结构。该优化在保持高C-O键氢解能力的同时,显著抑制了中间体的过度加氢,证明Co-FeOₓ协同作用增强了催化性能。
在含水共溶剂体系中,以FA为探针分子,我们通过动力学研究(包括同位素标记和动力学同位素效应(KIEs))探究了反应机制。这些实验揭示了水介质中溶剂调控的C-O键活化催化路径。多维表征(XRD、XPS、TEM、H₂-TPR)结合动力学研究建立了结构-性能关系,证明Fe诱导的电子效应降低了关键中间体(如氢化呋喃衍生物)的吸附强度,最终优化了反应路径。本研究为催化反应路径导向高附加值产物提供了新设计原则,同时为生物质分子增值利用的理性催化剂设计建立了理论框架。
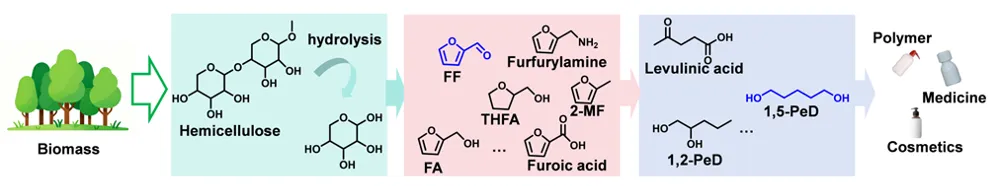
方案1 从木质纤维素生物质制备链状醇、羧酸和胺的生产路线
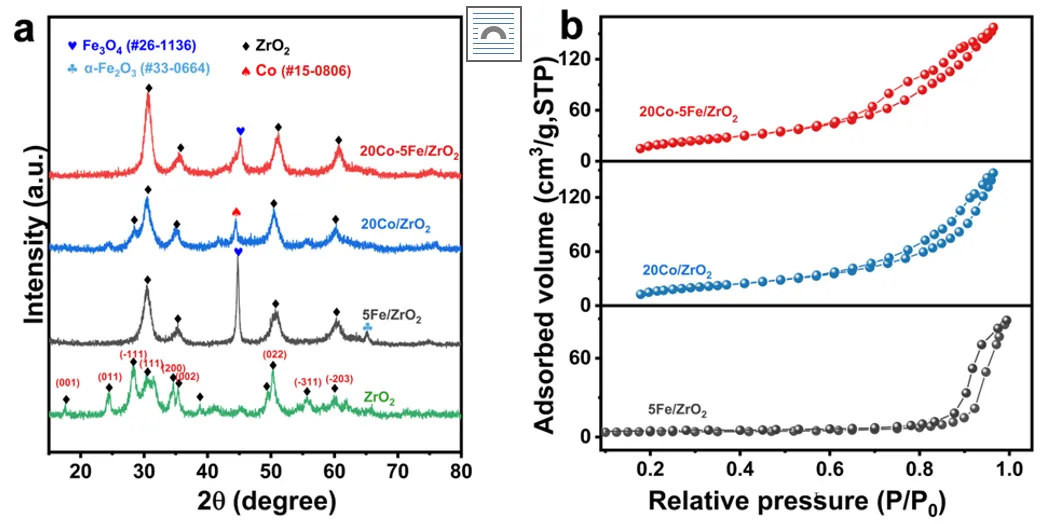
图1 (a) Co-Fe/ZrO₂催化剂的X射线衍射(XRD)图谱;(b) Co-Fe/ZrO₂催化剂的氮气吸附-脱附等温线。
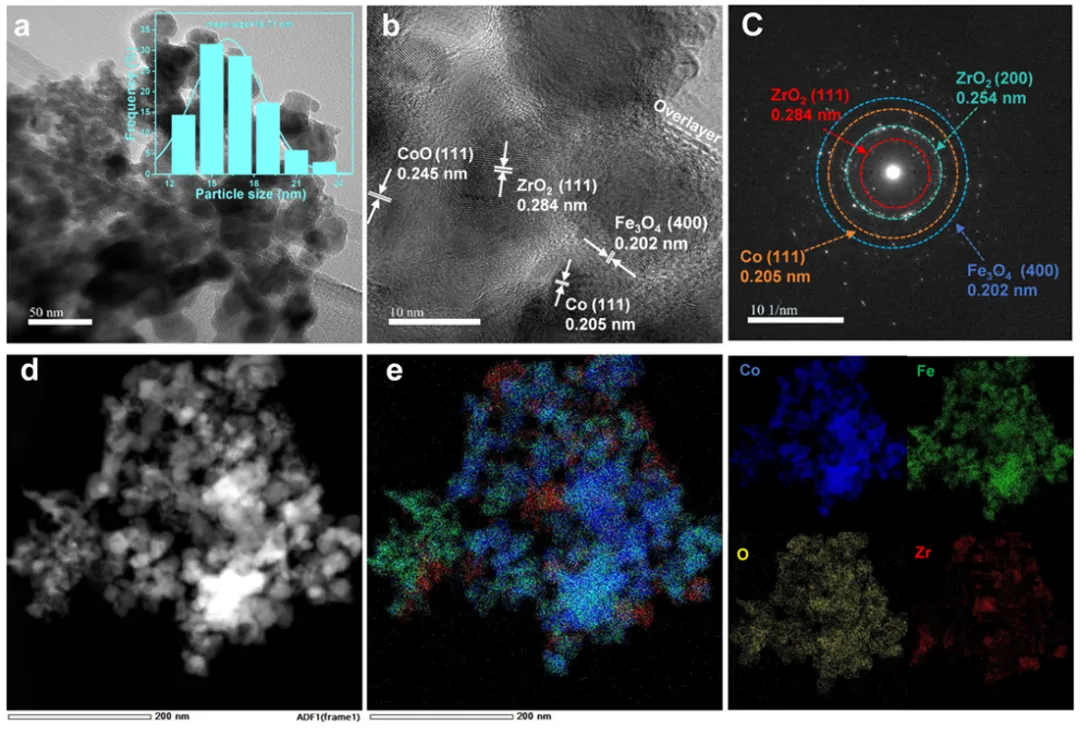
图2 (a,b) 20Co-5Fe/ZrO₂的高分辨率透射电子显微镜(HR-TEM)图像;(c) 快速傅里叶变换(FFT)图像;(d,e) Co-Fe/ZrO₂中钴(Co)、铁(Fe)和锆(Zr)的高角度环形暗场扫描透射电子显微镜(HAADF-STEM)图像及对应的能量色散X射线光谱(EDS)元素分布图。
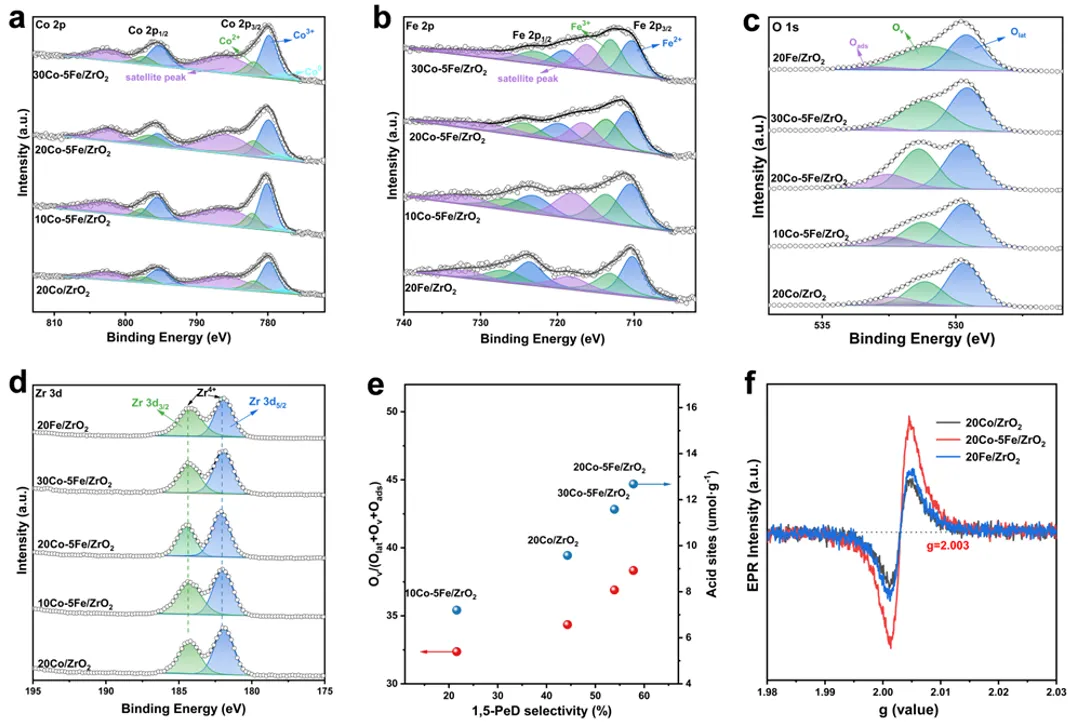
图3 Co-Fe/ZrO₂催化剂的X射线光电子能谱(XPS)图谱。(a) 钴(Co)2p轨道;(b) 铁(Fe)2p轨道;(c) 氧(O)1s轨道;(d) 锆(Zr)3d轨道;(e) 1,5-戊二醇(1,5-PeD)的选择性与Oᵥ/(Oₗₐₜ + Oᵥ + Oₐₛ)或酸性位点的关系;(f) Co-Fe/ZrO₂催化剂的X波段电子顺磁共振(EPR)图谱。
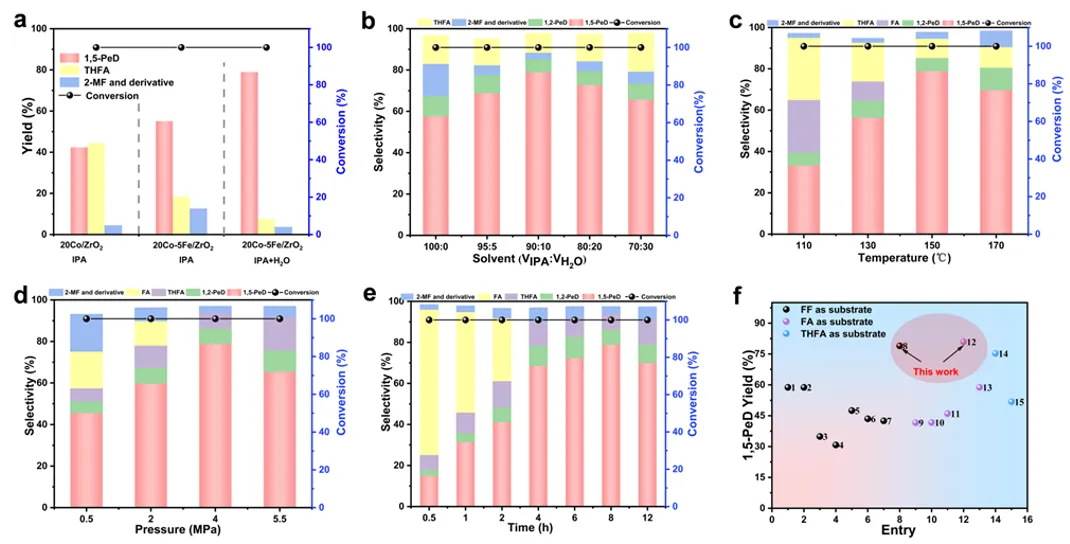
图4 20Co-5Fe/ZrO₂催化剂在不同条件下的糠醛(FF)转化率和产物选择性。(a) 在异丙醇/水(IPA/H₂O)体系中,20Co/ZrO₂和20Co-5Fe/ZrO₂催化剂上加氢产物的产率对比;(b) 反应条件:糠醛(1 mmol),催化剂(0.05 g),温度(150 ℃),氢气(H₂)压力(4.0 MPa),混合溶剂10 mL,反应时间(8 h);(c) 反应条件:糠醛(1 mmol),催化剂(0.05 g),氢气压力(4.0 MPa),混合溶剂(水/异丙醇,体积比10:90)10 mL,反应时间(8 h);(d) 反应条件:糠醛(1 mmol),催化剂(0.05 g),温度(150 ℃),混合溶剂(水/异丙醇,体积比10:90)10 mL,反应时间(8 h);(e) 反应条件:糠醛(1 mmol),催化剂(0.05 g),温度(150 ℃),氢气压力(4.0 MPa),混合溶剂(水/异丙醇,体积比10:90)10 mL;(f) 与文献中1,5-戊二醇产率的对比。
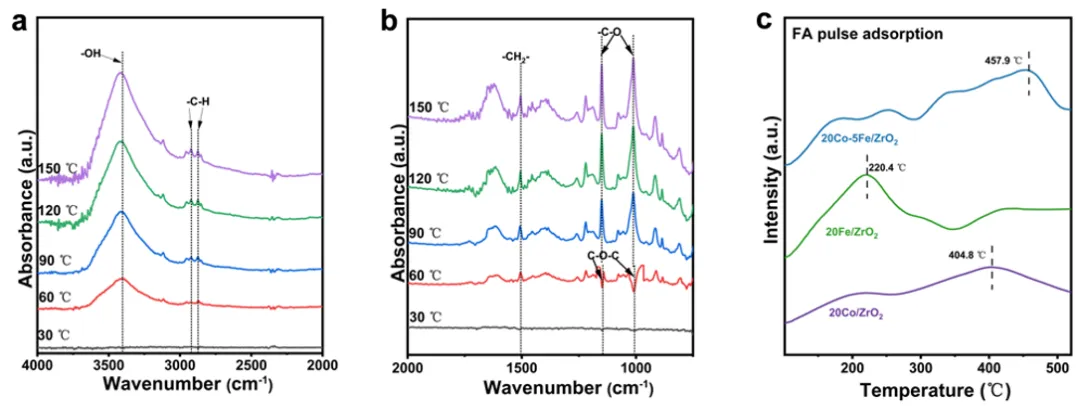
图5 (a, b) 20Co-5Fe/ZrO₂催化剂上糠酸(FA)加氢过程中的原位漫反射红外傅里叶变换光谱(DRIFT);(c) 20Co-5Fe/ZrO₂催化剂上糠酸的脉冲吸附。
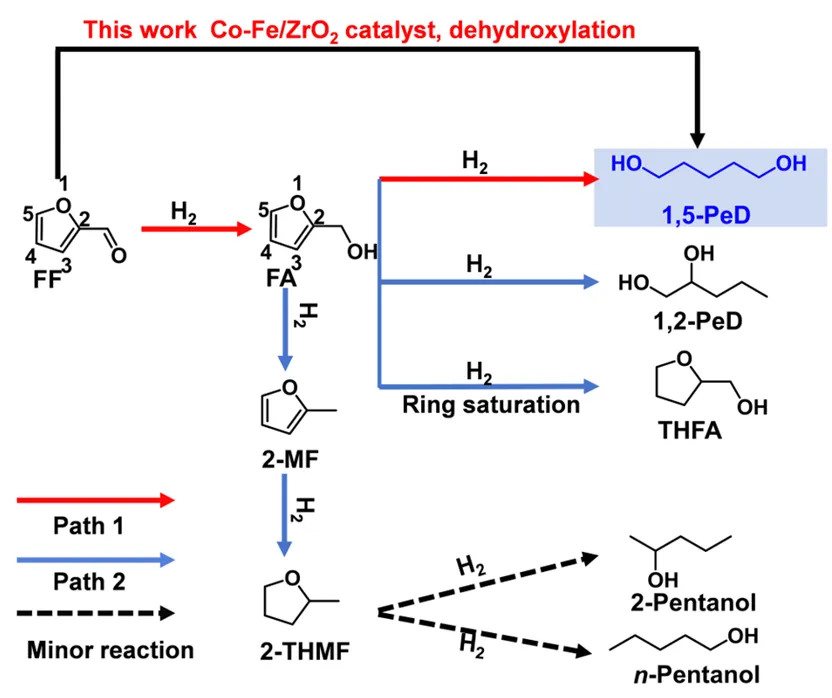 方案2 糠醛及其衍生物加氢反应网络
方案2 糠醛及其衍生物加氢反应网络
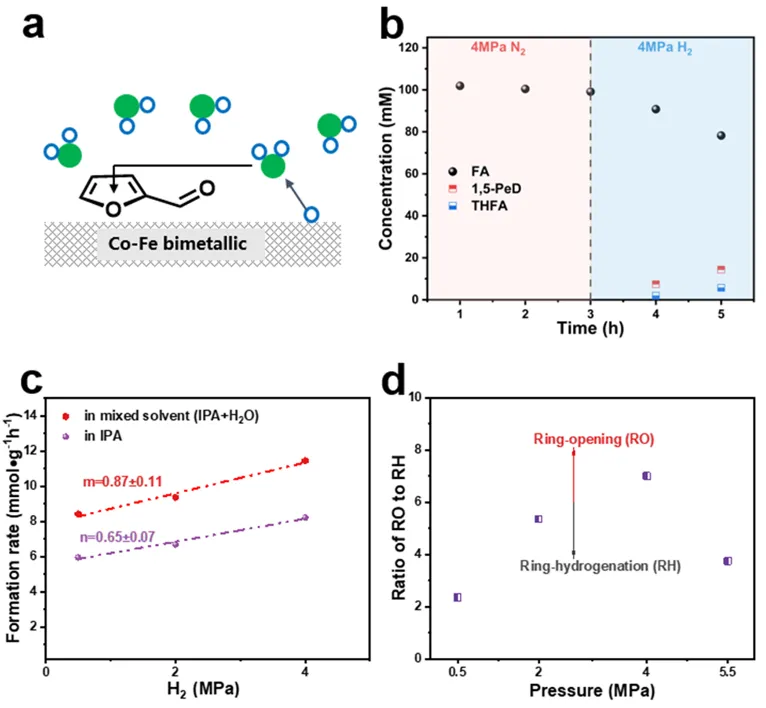
图6 (a) 类格罗特斯(Grotthuss-like)加氢机理的反应示意图;(b) 不同反应气氛下的对照实验。将反应混合物(糠酸、Co-Fe/ZrO₂、异丙醇)首先在氮气(N₂)气氛下于150 ℃加热3 h,然后在氢气气氛下再反应2 h;(c) Co-Fe/ZrO₂上关于氢气的反应级数;(d) 1,5-戊二醇和四氢糠醇(THFA)生成速率的比值。反应条件:糠醛(1 mmol),Co-Fe/ZrO₂(0.05 g),150 ℃,溶剂10 mL(水/异丙醇,体积比10:90)。
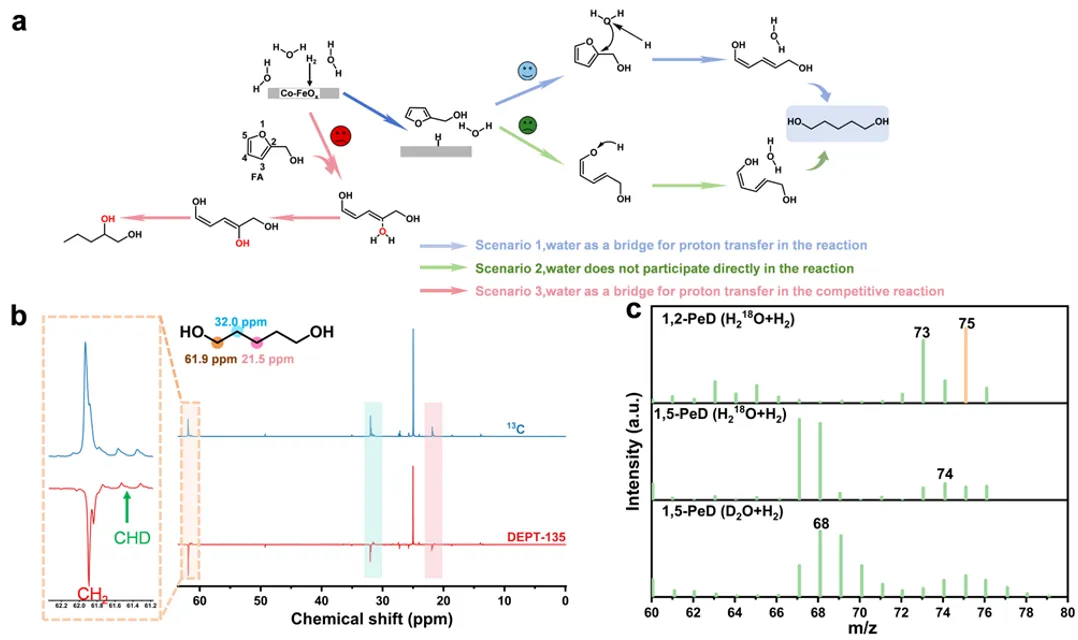
图7 (a) 水相中Co-FeOₓ表面上糠醛开环生成1,5-戊二醇的催化循环示意图。黑色箭头表示质子穿梭;(b) 由糠醛在重水(D₂O)中转化得到的1,5-戊二醇的碳-13核磁共振(¹³C NMR)和去耦碳-13核磁共振(DEPT135 NMR)图谱;(c) 由糠醛在含氧-18水(H₂¹⁸O)和重水(D₂O)中转化得到的1,2-戊二醇和1,5-戊二醇的质谱图。
我们开发了一种富氧()的钴-铁/氧化锆(Co–Fe/ZrO₂)催化剂,用于糠醛(FF)及其衍生物的开环加氢转化,以生产1,5-戊二醇(1,5-PeD)。与钴/氧化锆(Co/ZrO₂)催化剂相比,所提出的钴-铁/氧化锆(Co-Fe/ZrO₂)催化剂具有更低的活化能垒(Eₐ = 25.15 kJ/mol)、更强的C-O键断裂活性,并能抑制糠酸(FA)的过度加氢。结合实验和表征结果表明,铁的引入调节了钴位点的电子结构,降低了其对中间体的加氢亲和力,同时促进了动力学上较难进行的C-O键断裂——这是糠醛转化为1,5-戊二醇过程中的速率决定步骤。值得注意的是,通过糠酸加氢裂解实现了81%的1,5-戊二醇产率,这是迄今为止非贵金属基催化剂报道的最高值。同位素标记和动力学溶剂转换实验表明,金属表面吸附的氢物种的水诱导质子化机制通过质子耦合电子转移促进开环反应,绕过了直接由表面介导的步骤,并显著降低了C-O键断裂的活化能垒。本研究为从糠醛及其衍生物合成1,5-戊二醇提供了一种高性能的非贵金属催化剂。