基因鉴定:通过三种互补方法,共鉴定出15个推定的B型MtARR基因。
数据库验证:所有候选基因经数据库验证,确认为有效的家族成员(图S1)。
染色体分布:15个基因不均勻地分布于蒺藜苜蓿的6条染色体上,其中4号染色体上存在一个包含6个基因的密集簇,而5号和6号染色体上无分布(图S2;表S1)。
蛋白表征:编码蛋白的理化性质差异显著,分子量范围为25.36 kDa至75.52 kDa,等电点范围从酸性(4.88)到碱性(8.70)(表S2)。
亲疏水性:所有MtARR蛋白的亲水性平均值(GRAVY)均为负值,表明其为耐脱水蛋白。
亚细胞定位:预测所有15个MtARR蛋白均定位于细胞核内(表S2)。
通过结合基于AtARRs序列相似性的双向最佳匹配法、保守结构域鉴定和HMM模型筛选三种互补策略,在蒺藜苜蓿中鉴定出15个推定的B型MtARRs基因,并通过数据库验证确认为有效的家族成员,其数量与其他植物物种如紫花苜蓿(16个)和水稻(13个)相近(图S1)。这15个基因根据其染色体位置顺序命名为MtARR1至MtARR15,它们不均匀地分布在蒺藜苜蓿的6条染色体上,且基因数量与染色体长度不相关(图S2;表S1)。蛋白理化性质分析显示,其编码序列(CDS)长度和蛋白大小差异显著,分子量介于25.36 kDa至75.52 kDa之间,等电点(pIs)从酸性到碱性均有分布。所有MtARR蛋白的GRAVY值均为负,表明其为耐脱水蛋白,且均被预测定位于细胞核(表S2)。
3.2 B型MtARRs的系统发育进化分析与多序列比对
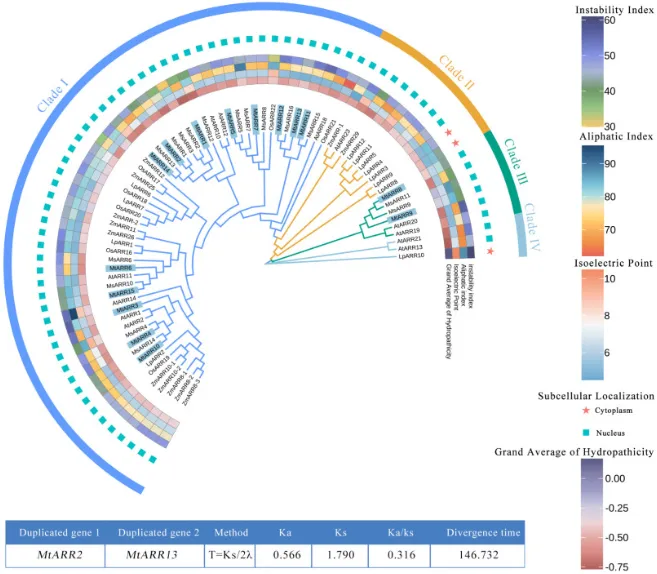
进化分析:构建了包含蒺藜苜蓿、紫花苜蓿、拟南芥等六个物种的系统发育树(图1;表S2)。
亚族分类:根据拟南芥的功能分类,15个MtARR成员被划分到四个主要分支中的两个,大部分属于分支I,MtARR8/9属于分支III,而分支II和IV中无MtARRs分布。
同源关系:MtARRs与其在另一豆科植物(紫花苜蓿)和单子叶植物中的同源蛋白关系比与拟南芥的更近。
序列相似性:MtARR与AtARR蛋白序列的相似性分析显示,部分潜在直系同源对具有较高的一致性,如AtARR1与MtARR10的相似性为64.82%(图S3)。
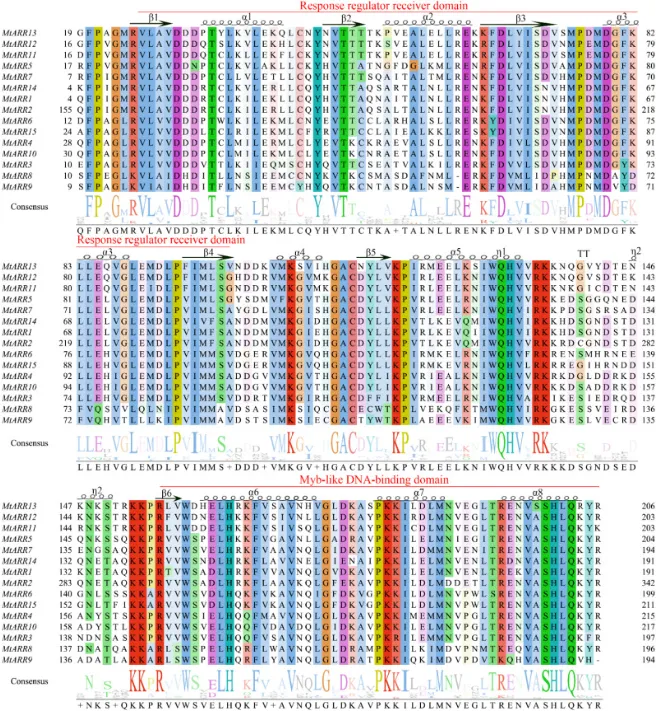
结构域保守:多序列比对显示,所有成员均含有高度保守的N端响应调控(REC)结构域和C端MYB样DNA结合结构域(图2)。
序列变异:连接功能结构域的接头区以及N端和C端区域在序列和长度上表现出显著的变异性。
为探究B型ARRs的进化关系,构建了包含蒺藜苜蓿(15个MtARRs)、紫花苜蓿(16个MsARRs)、多年生黑麦草(12个LpARRs)、玉米(12个ZmARRs)、水稻(7个OsARRs)和拟南芥(12个AtARRs)全长蛋白序列的系统发育树(图1;表S2)。根据B型AtARR基因家族的功能分类,该树状拓扑将蛋白分为四个主要分支,其中MtARR成员聚类于其中的两个分支,大部分属于分支I,MtARR8/9属于分支III,而分支II和IV中则没有MtARRs分布。通常,MtARRs与其在另一豆科植物(紫花苜蓿)和单子叶植物物种中的同源蛋白亲缘关系比与拟南芥的更近。此外,序列相似性矩阵分析揭示了MtARR和AtARR蛋白序列之间不同程度的一致性,其中一些潜在的直系同源对显示出较高的相似性,例如AtARR1和MtARR10之间的相似性为64.82%(图S3)。
图1. 跨多个植物物种的B型ARR蛋白的种间系统发育分析和理化性质表征。MtARRs的系统发育树是使用MEGA v7.0软件通过最大似然法(ML)构建的,并进行了1000次自举重复,包括来自蒺藜苜蓿、紫花苜蓿、拟南芥、玉米、水稻和多年生黑麦草的B型ARRs。中央蓝色背景表示MtARRs的成员,不同颜色的分支代表不同的B型ARRs亚家族,ARR的ID根据先前的研究命名。最外圈表示家族分类,不同的亚家族显示不同颜色的聚类分支。不同颜色的方形网格表示不同成员的理化性质和亚细胞定位位置。底部表格显示了重复基因的Ka/Ks值。
对15个家族成员的全长氨基酸序列进行系统性结构特征分析,结果显示,尽管它们的总长度差异显著,但所有成员均包含B型ARRs的两个特征结构域:一个高度保守的N端响应调控(REC)结构域和一个C端MYB样DNA结合结构域(图2)。此外,在几乎所有MtARRs中,C端MYB结构域附近都鉴定出一个高度保守的卷曲螺旋结构。对保守区域的深入分析表明,REC结构域内对磷酸中继至关重要的关键氨基酸残基,如天冬氨酸(D)磷酸化位点和负责稳定磷酸化状态的赖氨酸(K)残基,表现出高度的保守性。与这些功能域的高度保守形成鲜明对比的是,连接它们的接头区域以及N端和C端区域在序列和长度上均显示出显著的变异性。这种以保守功能域散布于可变区域为特征的结构组织表明,B型MtARR蛋白家族可能通过序列分化进化出了功能特异性,同时保留了其核心的信号转导和转录调控功能。
图2. B型MtARRs蛋白的多序列比对。 最保守的结构域用相同颜色和更高的一致性标记。α螺旋显示为黑色螺旋。延伸链显示为黑色箭头。“η”代表无规卷曲,“TT”代表β转角。
3.3 B型MtARRs的保守基序、结构域构架和基因结构
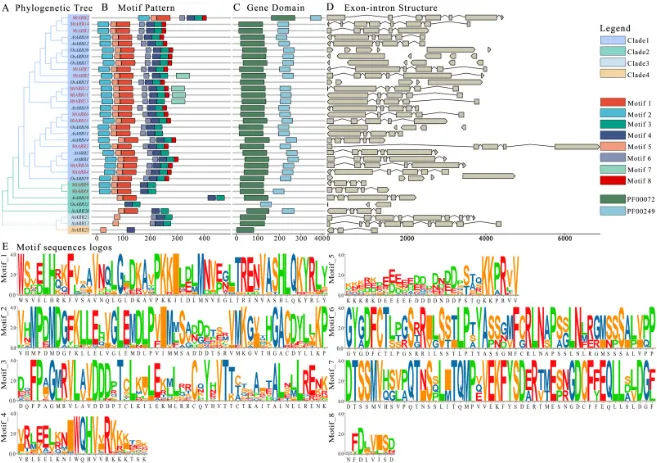
基序分析:在MtARR蛋白中鉴定出8个不同的保守基序,其中基序1、2、5共同对应REC结构域,而基序3和4对应MYB样DNA结合结构域(图3 A, B)。
结构域构架:所有15个MtARRs均包含典型的B型ARR结构,即一个N端响应调控(REC)结构域(PF00072)和一个C端MYB样DNA结合结构域(PF00249)(图3 C)。
基因结构:MtARR家族的内含子数量和长度表现出相当大的多样性,而同一系统发育分支内的基因则表现出非常相似的外显子-内含子模式(图3 D)。
基序保守性:序列标识图显示,构成这些基序的氨基酸残基在特定位置高度保守(图3 E)。
对15个B型MtARRs的保守基序、结构域构架和基因结构进行了分析(图3)。共鉴定出8个不同的基序,其中基序1、2和5共同对应于接收器REC结构域,而基序3和4对应于MYB样DNA结合结构域。这些基序的分布在同一系统发育分支的成员中高度一致,但在不同分支间则显示出显著差异(图3 A, B)。此外,所有15个MtARRs均包含典型的B型ARR结构,即一个N端响应调控(REC)结构域(PF00072)和一个C端MYB样DNA结合结构域(PF00249),证实了它们作为真正的B型ARR蛋白的身份(图3 C)。基因结构分析显示,内含子的数量和长度在MtARR家族中存在相当大的多样性,而同一亚族分支内的基因则表现出非常相似的外显子-内含子模式,进一步支持了进化分组的合理性(图3 D)。序列Logo显示,构成这些基序的氨基酸残基在特定位置高度保守(图3 E)。
图3. 蒺藜苜蓿中B型MtARRs家族成员的基序组成、保守结构域和基因结构分析。 (A) 使用邻接法和1000次自举重复构建的B型MtARRs种内系统发育关系,分支颜色区分不同进化枝。(B) 15个B型MtARRs的功能基序组成。8个基序由不同颜色的矩形框表示。(C) MtARRs中功能保守结构域PF00072和PF00249的组织结构,分别由深蓝色和深青绿色框表示。(D) MtARRs的内含子-外显子组织结构。内含子和外显子分别由断裂线和棕色框表示。(E) 已鉴定基序的序列标识图,图中彩色字母的大小代表相应氨基酸在该基序中的出现频率,字母越大,频率越高。
3.4 B型MtARRs的蛋白质二、三级结构预测分析
二级结构:MtARRs蛋白的二级结构主要由α-螺旋(15.81%–41.18%)和无规卷曲(39.37%–77.48%)组成,约占整个序列的80%(表S4)。
三级结构:所有MtARR蛋白显示出相似的整体空间构象,与二级结构预测高度一致(图S4)。
模型质量:基于SWISS-MODEL的同源建模结果显示,模型质量评估指标(GMQE为0.51–0.55,QMEANDisCo Global评分为0.36–0.42)表明MtARR蛋白三维结构模型质量较高(表S5)。
蛋白质结构分析显示,MtARRs蛋白的二级结构具有鲜明特征,主要由占整个序列约80%的α-螺旋(15.81%–41.18%)和无规卷曲(39.37%–77.48%)构成(表S4)。进一步的三级结构预测表明,所有MtARR蛋白均显示出相似的整体空间构象,其α-螺旋和无规卷曲在三维结构中的分布特征与二级结构预测高度一致(图S4)。基于SWISS-MODEL的同源建模结果显示,各目标蛋白与其最佳模板的序列相似性在70% - 82%之间,表明进化同源性高。模型质量评估指标(GMQE在0.51–0.55范围内,QMEANDisCo Global评分稳定分布在0.36–0.42范围内)表明,所构建的MtARR蛋白三维结构模型质量较高(表S5)。
3.5 B型MtARRs的基因复制与共线性分析
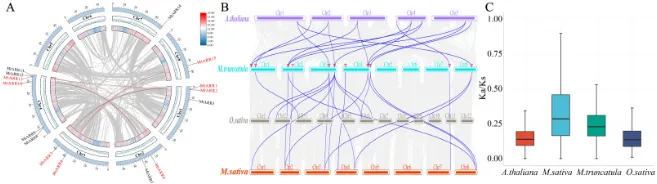
基因复制:种内共线性分析揭示了4对旁系同源基因(MtARR1/2、MtARR6/7、MtARR10/11和MtARR12/13),表明基因复制是该家族扩张的关键因素(图4 A)。
进化关系:种间共线性分析显示,蒺藜苜蓿与紫花苜蓿具有高度共线性,与拟南芥也存在大量直系同源对,而与水稻的共线性关系则很少,凸显了双子叶和单子叶植物之间的显著进化分歧(图4 B)。
选择压力:旁系和直系同源基因对的非同义替换率(Ka)与同义替换率(Ks)之比(Ka/Ks)均显著小于1.0,表明B型ARR基因在这些物种中经历了强烈的纯化选择压力(图4 C;表S6)。
为了探究MtARR基因家族的扩增机制,对15个B型MtARR基因的染色体分布和共线性进行了分析。在蒺藜苜蓿内部,种内共线性分析揭示了四对旁系同源基因,表明基因复制事件(可能是串联、全基因组或片段复制)是该家族进化的驱动力(图4 A)。为了将这些发现置于更广泛的进化背景中,进行了蒺藜苜蓿与三种代表性植物(拟南芥、水稻和紫花苜蓿)的比较性种间共线性分析。结果显示,蒺藜苜蓿与紫花苜蓿之间存在高度共线性,与拟南芥也存在大量直系同源对,而与水稻的共线性关系则非常有限,这凸显了双子叶和单子叶植物谱系之间的显著进化分歧(图4 B)。对这些基因的选择压力评估显示,所有旁系和直系同源基因对的Ka/Ks比值均显著小于1.0,表明B型ARR基因在这些物种的进化过程中受到了强烈的纯化选择(图4 C;表S6)。
图4. B型ARR基因的染色体分布与共线性分析。 (A) 蒺藜苜蓿中B型ARRs的染色体定位和种内共线性的环形图。基因分布在六条染色体(Chr1 – Chr6)上,共线基因对由灰色线连接。红色基因名表示存在共线关系,而黑色名称代表非共线基因。内环代表每条染色体的基因密度梯度。(B) 蒺藜苜蓿与3种代表性植物(拟南芥、水稻和紫花苜蓿)之间B型ARRs的共线性分析。背景中的灰色线表示蒺藜苜蓿和其他植物基因组中的共线区块,而蓝色线则突出显示了同源的ARR基因对。(C) 四个物种中直系同源和旁系同源基因对的Ka/Ks值分布,显示了纯化选择压力(Ka/Ks < 1.0)。误差棒代表标准差,方框内的水平线表示中值。
3.6 B型MtARR启动子的顺式作用元件分析
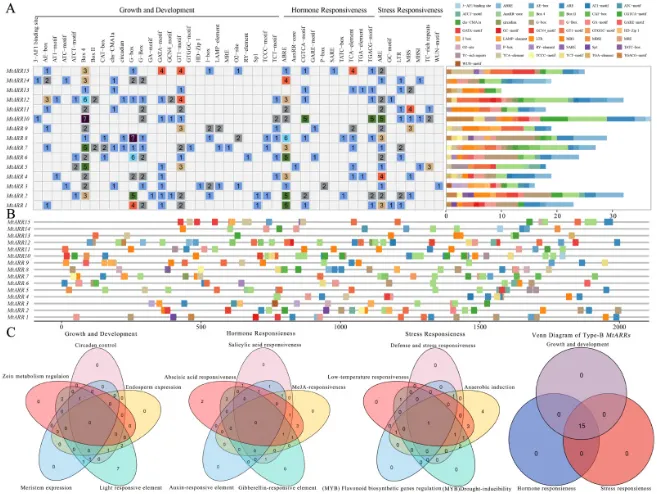
元件鉴定:在15个MtARR基因的启动子区域共鉴定出43类、348个独立的顺式作用元件(表S7)。
功能分类:这些元件被分为三类:生长发育元件、激素响应元件和胁迫响应元件。
元件丰度:与光响应(G-Box和Box4)、脱落酸响应(ABRE)和无氧响应(ARE)相关的元件最为丰富。
分布特征:元件在启动子区域的分布不均匀,在转录起始位点上游的近端区域(约前500 bp)密度更高(图5 B)。
调控复杂性:所有15个基因均包含三大功能类别的元件,且不同元件的组合和数量各异,如MtARR10拥有最丰富的元件类型和数量,暗示了复杂精密的调控网络(图5 A, C)。
为探究B型MtARR基因的潜在调控机制,对其转录起始位点上游2000 bp的启动子区域进行了分析(图5)。共鉴定出43种、总计348个顺式作用元件,这些元件可分为三大功能类别:(1) 生长发育元件,(2) 激素响应元件,(3) 胁迫响应元件(表S7)。其中,与光响应(G-Box、Box4)、脱落酸响应(ABRE)和无氧响应(ARE)相关的元件最为丰富。这些元件在启动子区域的分布不均匀,通常在转录起始位点上游的近端区域(约前500 bp)呈现更高的密度(图5 B)。每个基因的元件数量和类型差异显著,其中MtARR10拥有的元件数量最多(图5 A)。维恩图分析进一步揭示了调控功能的重叠与特异性,所有15个B型MtARR基因均包含三大功能类别的元件,表明它们在协调这些不同生物信号中发挥着整合作用(图5 C)。综上所述,B型MtARR基因启动子中顺式作用元件的多样化组成、数量和分布,暗示了一个复杂而精密的调控网络。
图5. 蒺藜苜蓿中B型ARRs基因家族顺式作用元件的综合分析。 (A) 每个MtARRs中顺式作用元件的数量和功能分类。图中展示了在每个MtARRs中鉴定出的43种顺式作用元件的分布;元件由不同颜色的方框表示。(B) 所有15个MtARR基因2000 bp启动子区域内顺式作用元件的定量分布,显示了位置偏好和聚类模式。综合图例用颜色编码显示了所有43种元件类型。(C) MtARRs的维恩图,展示了三大功能类别(生长/发育、激素信号和胁迫响应通路)中顺式调控元件的重叠和特异性,揭示了调控的复杂性和潜在的串扰。
3.7 B型MtARR基因的时空表达谱
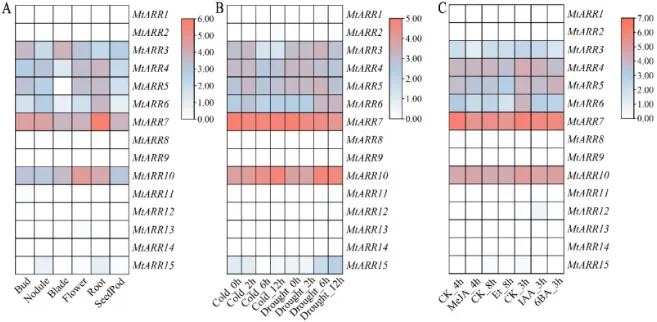
组织特异性:公共转录组数据显示,15个MtARR基因在六种发育组织中表现出显著不同的表达模式,如MtARR7在根中特异性高表达,MtARR10在花中高表达(图6 A)。
胁迫响应:在干旱胁迫下,包括MtARR3/4/5/6/7/10在内的一组基因显著上调(图6 B)。
激素响应:MtARR7受细胞分裂素(CK)显著诱导,而MtARR4/5/7/10等一组基因则被茉莉酸甲酯(MeJA)强烈上调(图6 C)。
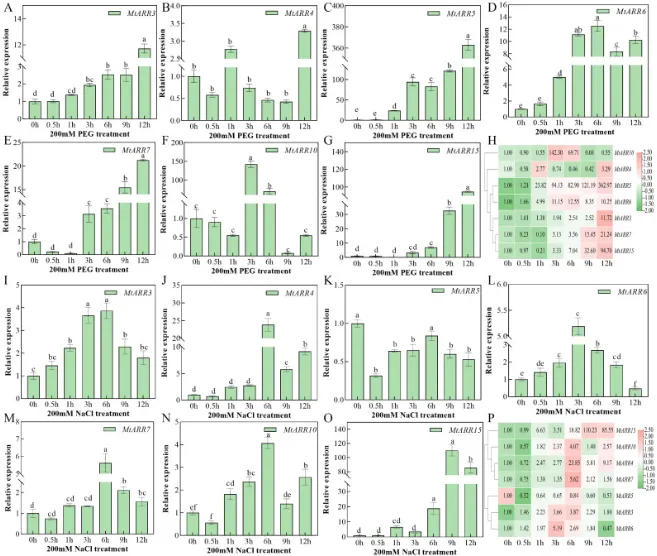
表达验证:RT-qPCR结果验证了所选基因在不同组织中的广泛表达和时空表达差异,并根据表达模式将其分为三类(图7)。
胁迫表达动态:在干旱、盐和低温胁迫下,所选MtARRs基因的表达水平均表现出动态变化。值得注意的是,MtARR4和MtARR10在干旱和盐胁迫下的表达趋势高度相似(图8;图S5)。
激素表达动态:在多种激素(ETH、ABA、SA、MeJA、IAA、6-BA)处理后,MtARRs基因均表现出响应,但响应模式各异,暗示它们在不同激素信号转导通路中可能扮演不同角色(图S6)。
为阐明B型MtARR基因的潜在功能,利用公共转录组数据分析了它们在不同组织、非生物胁迫和激素处理下的表达谱,结果揭示了高度异质化的模式(图6,表S8&9)。在组织特异性表达方面,MtARR7在根中表现出极高的表达,而MtARR10在花中高表达(图6 A)。在非生物胁迫条件下,一组基因(包括MtARR3/4/5/6/7/10)在干旱胁迫下显著上调(图6 B)。激素诱导的表达变化显示,MtARR7被细胞分裂素(CK)急剧诱导,而一组基因(包括MtARR4/5/7/10)则被茉莉酸甲酯(MeJA)强烈上调(图6 C)。
图6. MtARR基因在不同条件下的时空表达谱分析。 (A) 在六个发育阶段/组织中的组织特异性表达模式:芽、根瘤、叶片、花、根和豆荚。表达值经过log₂转换,并以蓝到红的色阶表示低到高表达水平的热图呈现。(B) 在非生物胁迫条件(4°C低温和干旱胁迫)下多个时间点(0小时、2小时、6小时、12小时)的时间表达动态。数据来源于蒺藜苜蓿基因表达图谱,显示了家族成员间的差异性胁迫响应。(C) 经六种植物激素处理后的激素诱导表达变化:细胞分裂素(CK)、茉莉酸甲酯(MeJA)、赤霉酸(GA)、乙烯(ET)、吲哚-3-乙酸(IAA)和6-苄基氨基嘌呤(6-BA),在不同时间间隔进行。表达数据经过归一化和聚类,以揭示激素特异性响应模式和潜在的调控网络。
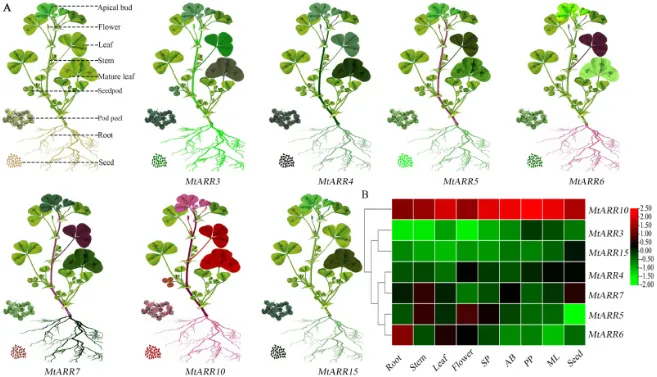
RT-qRCR进一步验证了MtARR3/4/5/6/7/10/15在蒺藜苜蓿不同组织中的表达模式,结果表明这些基因广泛表达但具有不同的时空表达模式,并可大致分为三类(图7 A, B)。
图7. 选定的MtARR基因(MtARR3、MtARR4、MtARR5、MtARR6、MtARR7、MtARR10和MtARR15)在九个不同蒺藜苜蓿器官(根、茎、叶、花、SP、AB、ML和种子)中的组织特异性表达验证和可视化。 SP,种皮;AB,顶芽;ML,成熟叶。每个组织中的颜色强度对应于通过qRT-PCR分析确定的相对表达水平,并带有组织特异性注释:顶芽、花、叶、茎、成熟叶、豆荚、荚果皮、根和种子。(B) qRT-PCR验证的表达数据的层次聚类热图,显示了基于组织特异性模式的三个不同表达组。
在胁迫处理下,MtARR3/5/7/15的表达水平在干旱胁迫下普遍上调。在盐胁迫下,MtARR4和MtARR10的表达急剧增加后下降,随后在后期再次上升,且两者在干旱和盐胁迫下的表达趋势高度相似(图8)。在低温胁迫下,MtARRs基因的表达整体呈现先降后升再回落的趋势(图S5)。在多种激素处理后,MtARRs的表达也发生了显著变化,例如MtARR4/6/10在ABA诱导下显著增加,表明它们可能在ABA信号转导中发挥重要作用(图S6)。
图8. MtARR基因在非生物胁迫条件下的时间表达谱。 七个代表性MtARR基因(MtARR3/4/5/6/7/10/15)在(A-G) 200 mM聚乙二醇(PEG)处理诱导的干旱胁迫和(I-O) 200 mM NaCl处理诱导的盐胁迫下,在12小时时间进程(0、0.5、1、3、6、9和12小时)中的定量RT-PCR分析。相对表达水平使用2-ΔΔCt方法计算,以MtActin为参考基因,并对0小时对照进行归一化。误差棒代表三个生物学重复的标准误。条形图上方的不同字母表示统计学上的显著差异(p < 0.05,单因素方差分析后进行邓肯多重范围检验)。(H, P) 分别显示了七个MtARR基因在干旱和盐胁迫条件下的共表达关系的相关性矩阵。颜色强度和数值代表皮尔逊相关系数,红色表示正相关,绿色表示负相关。
3.8 亚细胞定位与转录活性分析
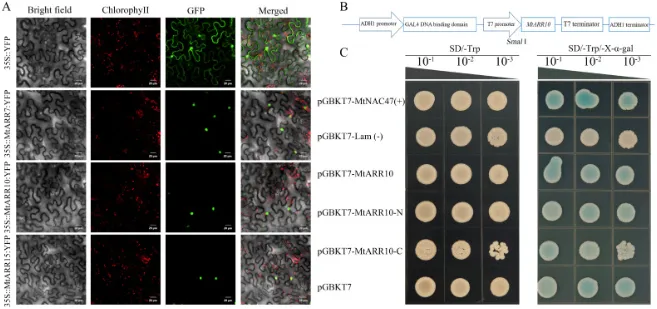
亚细胞定位:生物信息学预测和实验验证均表明,MtARR7、MtARR10和MtARR15均为核定位蛋白,这与其作为转录调控因子的潜在作用一致(图9 A)。
转录自激活:利用GAL4系统的酵母实验证实,MtARR10蛋白具有很强的转录自激活活性(图9 C)。
激活域分析:MtARR10的全长、N端和C端片段均能激活报告基因,表明其N端和C端区域内均可能存在独立的激活域(图9 B, C)。
为了确定MtARR7/10/15蛋白的亚细胞定位,首先通过生物信息学工具预测其为核蛋白。随后,通过将其全长编码序列与绿色荧光蛋白(GFP)融合,并在植物细胞中瞬时表达,实验性地验证了这一预测。荧光显微镜观察显示,35S::MtARR7-YFP、35S::MtARR10-YFP和35S::MtARR15-YFP的绿色荧光信号均专一地定位于细胞核内(图9 A)。为确定MtARR10蛋白是否具有转录自激活活性,进行了基于酵母GAL4系统的实验。结果显示,表达全长MtARR10及其独立的N端和C端结构域的酵母均能在报告培养基上变蓝,表明MtARR10具有很强的转录自激活活性,并且该功能可能由其N端和C端区域内的不同激活域介导(图9 B, C)。
图9. 亚细胞定位与转录激活。 (A) 3个MtARRs蛋白在烟草叶片中的亚细胞定位。该图显示了明场、叶绿素自发荧光、GFP荧光以及合并通道的荧光显微镜图像(从左到右)。比例尺代表20 μm。(B) pGBKT7-MtARR10重组载体的构建示意图,显示了MtARR10基因在Smal I限制性位点后的插入情况。(C) MtARR10蛋白在酵母细胞中的转录激活分析。将不同载体转化到酵母中,并在色氨酸缺陷培养基(SD/−Trp)和含有X-α-gal的选择性培养基(SD/−Trp/−X-α-gal)上生长。载体如下:pGBKT7-MtNAC47,阳性对照(+);pGBKT7-Lam,阴性对照(−);pGBKT7,空载体;pGBKT7-MtARR10,全长蛋白;pGBKT7-MtARR10-N,N端结构域(氨基酸1–1023);pGBKT7-MtARR10-C,C端结构域(氨基酸1024–2046)。
3.9 通过酵母过表达系统观察胁迫响应
功能验证:通过在酿酒酵母中异源表达MtARR基因,评估其在非生物胁迫响应中的作用。
正向调控:转化了pYES2-MtARR7/10的酵母菌株在盐胁迫(高浓度NaCl)和渗透胁迫(高浓度PEG6000)下,均表现出比空载体对照显著增强的耐受性(图10)。
负向调控:相比之下,表达pYES2-MtARR15的菌株在盐胁迫下耐受性没有改善,而在渗透胁迫下则表现出超敏表型,生长受到显著抑制(图10)。
功能分化:这些结果有力地表明,这些MtARR基因在响应非生物胁迫方面存在功能分化,MtARR7和MtARR10作为盐和渗透胁迫耐受性的正调控因子,而MtARR15则作为负调控因子。
为了功能性地验证MtARR基因在非生物胁迫响应中的作用,评估了在盐和渗透胁迫条件下转化了pYES2-NTB(空载体对照)、pYES2-MtARR7/10/15的酿酒酵母INVSC1菌株的生长特性。在盐胁迫下,转化了pYES2-MtARR7/10的酵母菌株表现出显著增强的耐受性。相反,表达pYES2-MtARR15的菌株则未显示出盐耐受性的改善(图10 A)。在由PEG6000模拟的渗透胁迫下,携带pYES2-MtARR7/10的菌株表现出优于对照的生长性能。相反,pYES2-MtARR15的表达导致了超敏表型,其生长受到显著抑制(图10 B)。这些结果强烈暗示了这些MtARR基因在响应非生物胁迫方面存在功能分化:MtARR7和MtARR10作为盐和渗透胁迫耐受性的正调控因子,而MtARR15则似乎作为负调控因子。
图10. 通过酵母异源表达对MtARRs胁迫耐受性的功能验证。 转化了pYES2-NTB(空载体对照)或pYES2-MtARRs(MtARR7、MtARR10、MtARR15)的酿酒酵母INVSC1菌株在(A)盐胁迫条件(0–5 M NaCl)和(B)渗透胁迫条件(0–45% PEG6000)下的生长性能。酵母培养物经过系列稀释(10⁻⁴、10⁻⁵、10⁻⁶)后,点样于补充了半乳糖以诱导蛋白表达的SD/-Ura选择性培养基上。