

锂硫电池(LSB)因其高能量密度和理论比容量,被视为极具前景的下一代储能系统。然而,其硫氧化还原反应中的准液-固转化步骤(Li₂S₄ → Li₂S)被公认为是整个反应的速率决定步骤(RDS)。该步骤动力学极其缓慢,不仅贡献了四分之三的理论容量,其迟缓的速率还导致多硫化物中间体积累,引发严重的穿梭效应,成为限制LSB性能的关键瓶颈。因此,如何有效加速这一决速步的动力学,是提升LSB整体性能的核心挑战。
2026年5月18日,河南农业大学吴俊锋教授、郑州大学唐宾副研究员、南开大学周震教授在《Advanced Materials》上发表了题为“Solid-Liquid Synergy Enables a Trisulfur-Radical-Rich Microenvironment for Accelerated Li-S Conversion Kinetics”的研究论文。该工作提出了一种创新的固-液协同策略,通过集成富含氧空位的Co掺杂Fe₂O₃(Co-Fe₂O₃)硫宿主与作为电解液添加剂的1,3-二甲基-2-咪唑啉酮(DMI),在正极侧构建了一个富含三硫自由基(S∙⁻₃)的局部高给体数(高DN)微环境。
研究团队首先通过实验和理论计算证实,Co-Fe₂O₃中由Co掺杂诱导的大量氧空位,对高DN溶剂DMI分子中的羰基(C=O)具有强化学吸附作用。开尔文探针力显微镜(KPFM)结果显示,经DMI处理的Co-Fe₂O₃表面接触电位差显著升高,证明DMI优先富集于催化剂表面,成功形成了与体相电解液不同的局部高DN微环境。原位拉曼光谱和紫外-可见光谱证实,在该微环境中,高DN溶剂有效稳定了高活性的S∙⁻₃中间体。DFT计算进一步揭示,S∙⁻₃介导的Li₂S₄转化路径(Li₂S₄ + LiS∙₃ + 11Li⁺ → 7Li₂S)具有更低的吉布斯自由能变化(-405.04 kcal mol⁻¹),远优于传统路径(-205.97 kcal mol⁻¹),从热力学上证实了其加速决速步的能力。
得益于S∙⁻₃对RDS的显著加速作用,该体系在电化学性能上表现卓越。组装的LSB在1C倍率下循环500圈后,容量保持率高达85.4%,平均每圈衰减率仅0.03%。其倍率性能同样出色,在5C的高电流密度下仍能释放出659.6 mAh g⁻¹的初始容量。即使在4.6 mg cm⁻²的高硫载量下,也能实现1126.9 mAh g⁻¹的高初始容量。恒电位沉积实验和扫描电镜表明,S∙⁻₃的介入促进了三维Li₂S的沉积,形成了疏松多孔的结构,避免了传统二维致密沉积层导致的电极钝化,进一步提升了反应可逆性。变温阻抗分析表明,该策略将决速步的表观活化能(Ea₂)从69.2 kJ mol⁻¹降低至65.8 kJ mol⁻¹,对应室温下反应速率常数提升约3.9倍,为动力学优化提供了直接证据。

1、通过富氧空位的Co-Fe₂O₃优先吸附高DN溶剂DMI,在正极界面创建独特微环境,有效稳定高活性三硫自由基。
2、实验与理论计算共同证实,S∙⁻₃作为关键介体,从根本上加速了Li₂S₄到Li₂S的决速步,显著提升转化动力学。
3、受益于RDS的加速,LSB在5C高倍率下容量达659.6 mAh g⁻¹,并在1C下循环500圈后容量保持率高达85.4%。

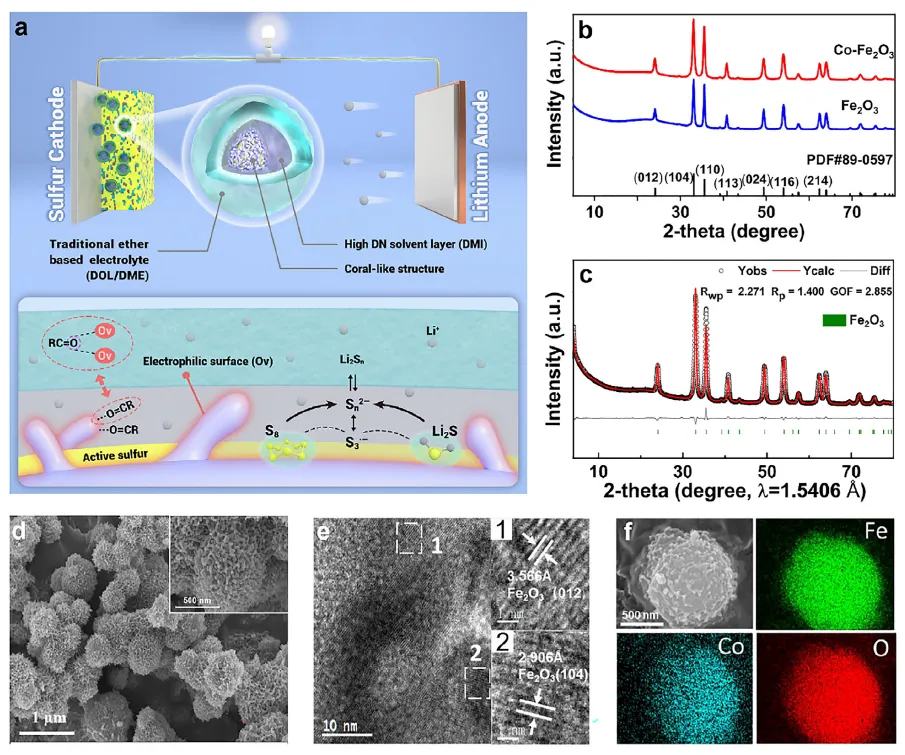
图1 固-液协同策略与催化机制示意图及Co-Fe₂O₃材料表征
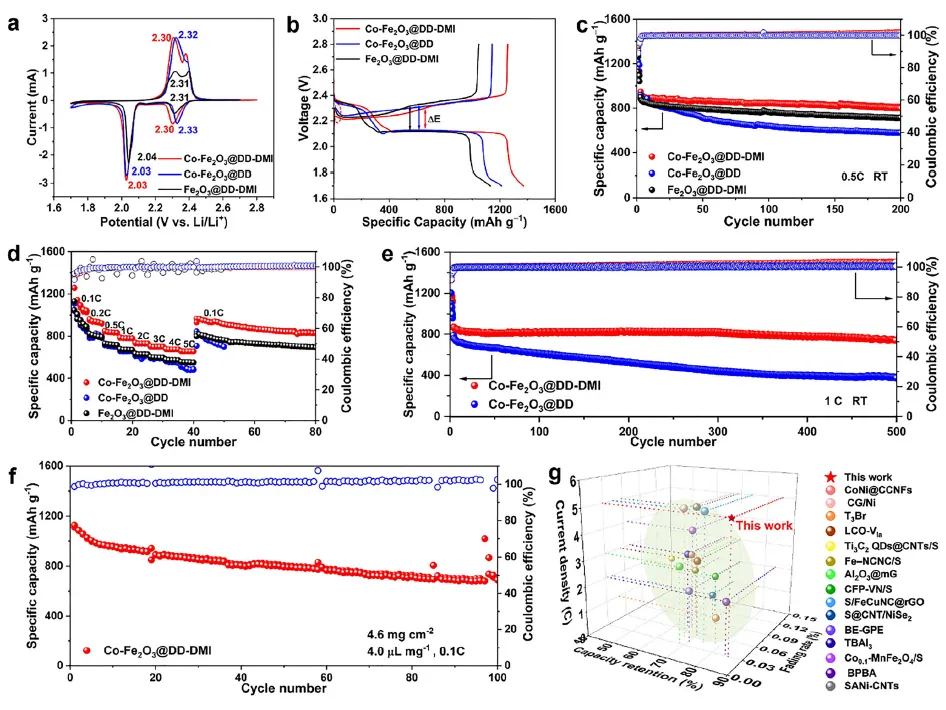
图2 电化学性能
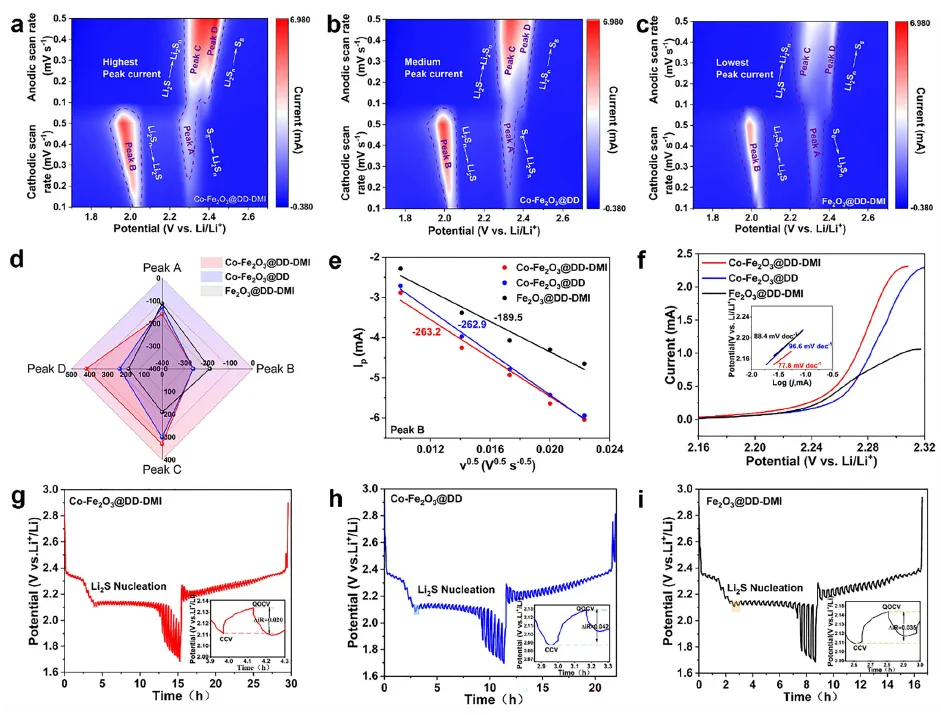
图3 反应动力学分析
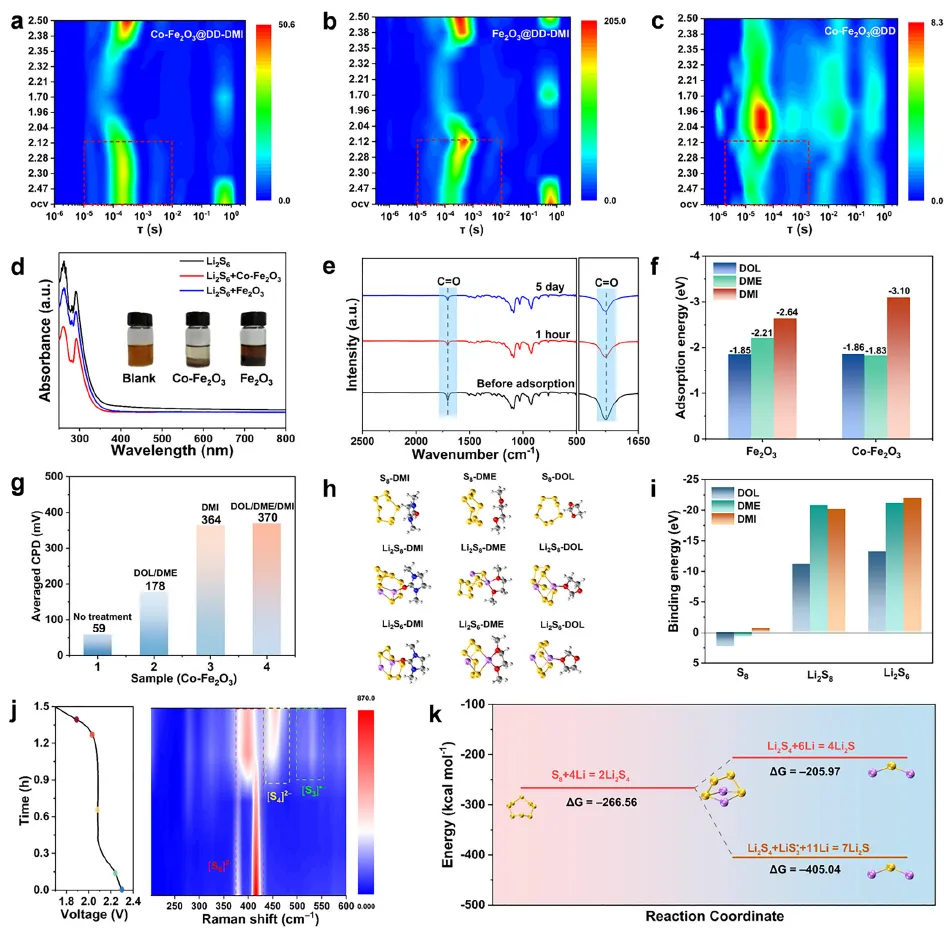
图4 锂硫电池中的吸附效应
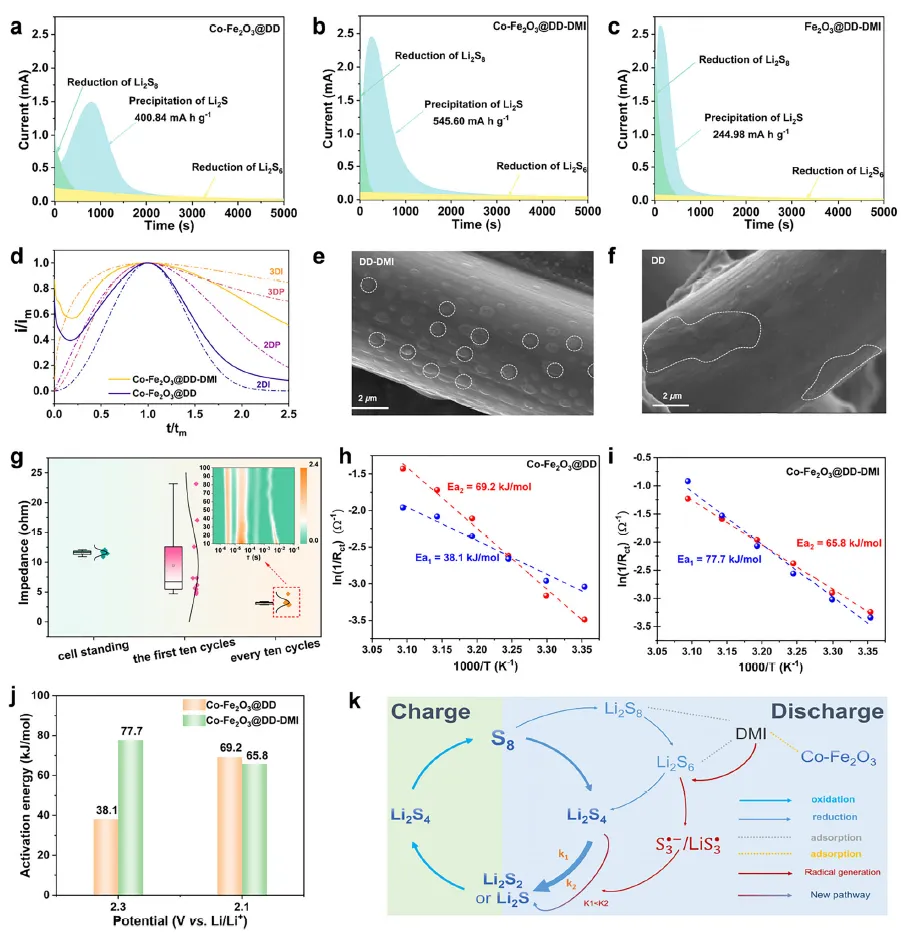
图5 Li₂S沉积行为

本研究成功开发了一种固-液协同策略,通过将富含氧空位的Co-Fe₂O₃宿主与高DN溶剂DMI添加剂相结合,在锂硫电池正极构建了局部高DN微环境。该环境有效稳定了三硫自由基(S∙⁻₃),并证实其作为关键介体,从热力学和动力学上加速了S的氧化还原反应中从Li₂S₄到Li₂S的决速步。这项工作揭示了一种全新的S∙⁻₃介导催化机制,通过界面工程与电解液调制的协同,突破了决速步固有的动力学限制,为开发高比能、长寿命锂硫电池提供了全新的思路和坚实的理论依据。
文献链接:https://doi.org/10.1002/adma.73407
注:本文仅供科研人员学术交流使用,未涉及任何商业用途。由于小编水平有限,文中如有不当或不准确之处,敬请批评指正。如有疑问,欢迎随时联系小编交流探讨。

 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
