①基于结构的设计策略
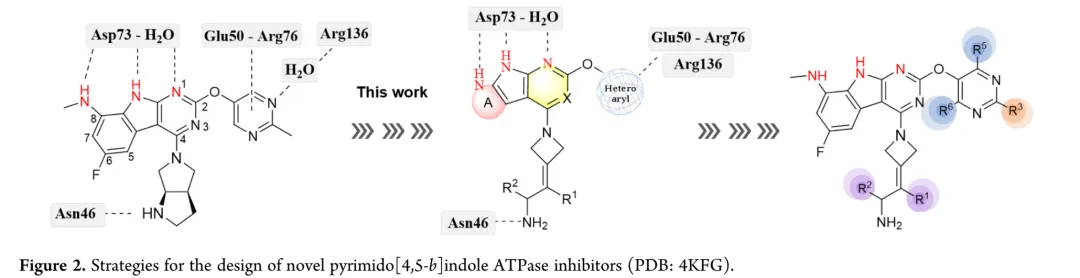
吡啶并[4,5-b]吲哚骨架与大肠杆菌GyrB的ATP结合位点形成关键氢键网络,包括与Asp73羧酸基团及结构保守水分子的相互作用,C-2嘧啶部分与Glu50-Arg76盐桥形成π-阳离子相互作用,同时与Arg136形成水介导氢键,而与Asn46通过水网络形成的氢键相互作用与抗革兰氏阴性菌活性密切相关。化合物8在大鼠肝微粒体中的代谢物分析显示,C-8位去甲基化(代谢物10)是主要代谢途径,占总检测代谢物的73%,该代谢物丧失抗菌活性,解释了尽管化合物9体外活性强但体内疗效减弱的原因;Kong等采用氘代甲基(-CD3)替代C-8甲基以阻断代谢,但意外的是氘代反而降低系统暴露量并增加清除率,表明代谢并未改善。基于这些发现,本研究的初始优化策略在保留关键药效团相互作用的同时修饰核心骨架:通过环化策略将代谢不稳定的C-8甲基纳入吡唑环,并评估对抗菌活性的影响;主要策略是实施骨架杂化,将Roche的吡啶并[2,3-b]吲哚与吡啶并[4,5-b]吲哚亚结构结合以评价活性后果;进一步修饰探索C-2位的芳香杂环,以维持与Glu50-Arg76的π-阳离子相互作用,同时用水介导接触替换为与Arg136的直接氢键;基于计算机辅助药物设计结果,认识到修饰R3位点的合理性,进而开展系统的构效关系研究;R3和R4位点的综合修饰成功阐明了吡啶并[4,5-b]吲哚衍生物的构效关系(STR)和构代关系(SMR),该整合策略实现了活性、hERG抑制和药代动力学性质的平衡,最终在小鼠耐多药鲍曼不动杆菌感染模型中验证了疗效。
②从骨架修饰到计算机辅助药物设计指导的强效类似物发现
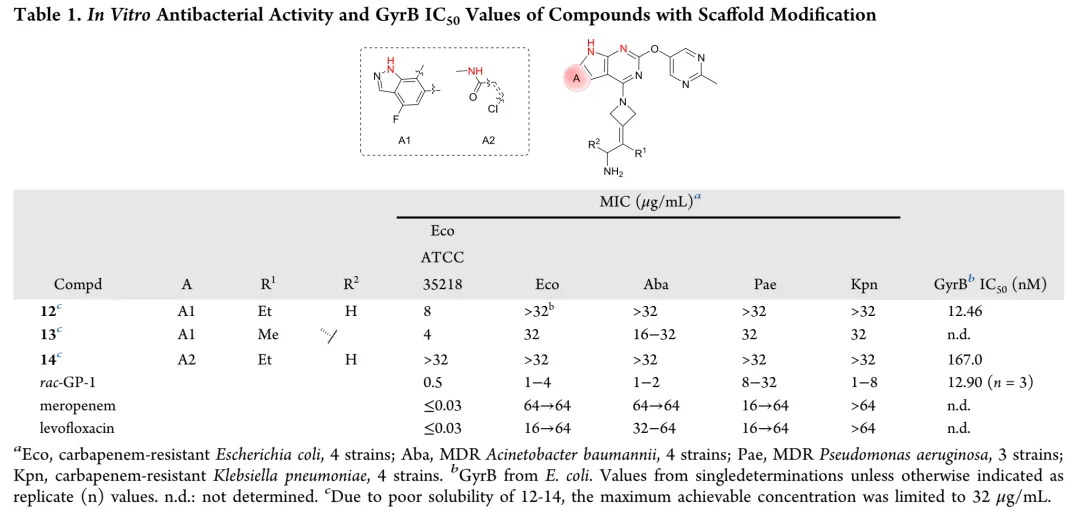
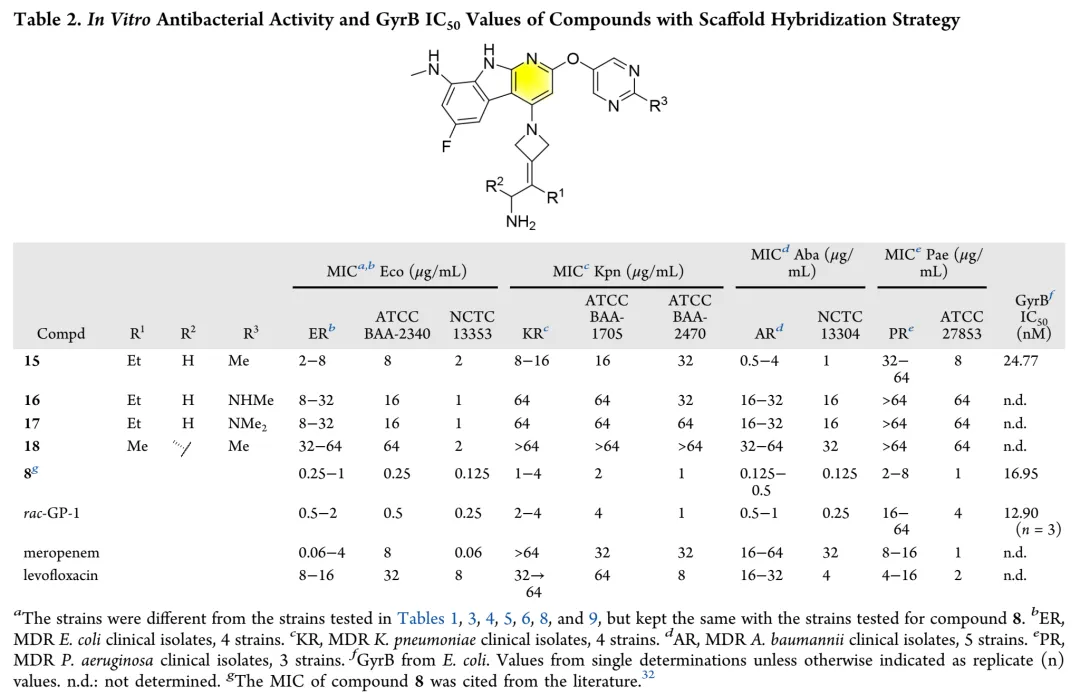
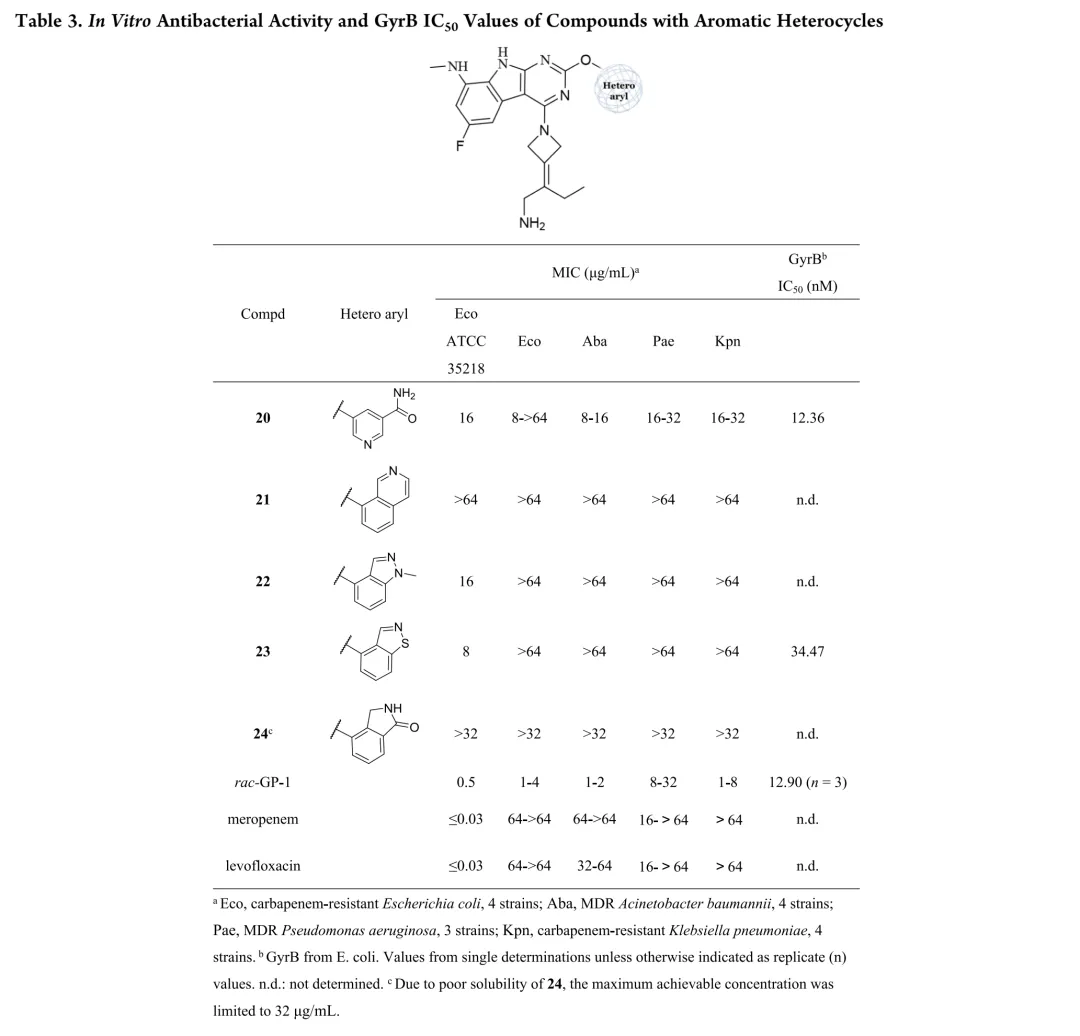
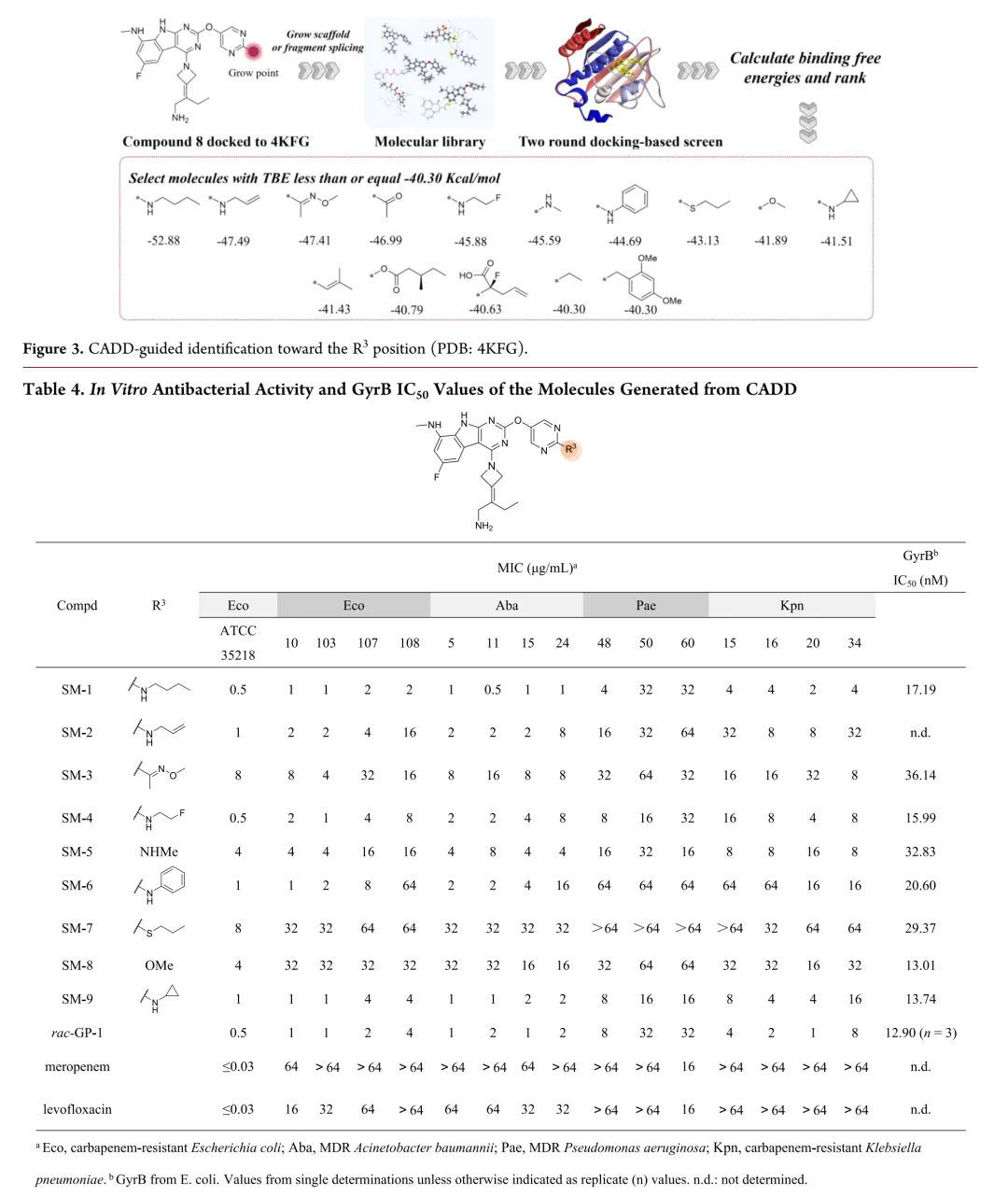
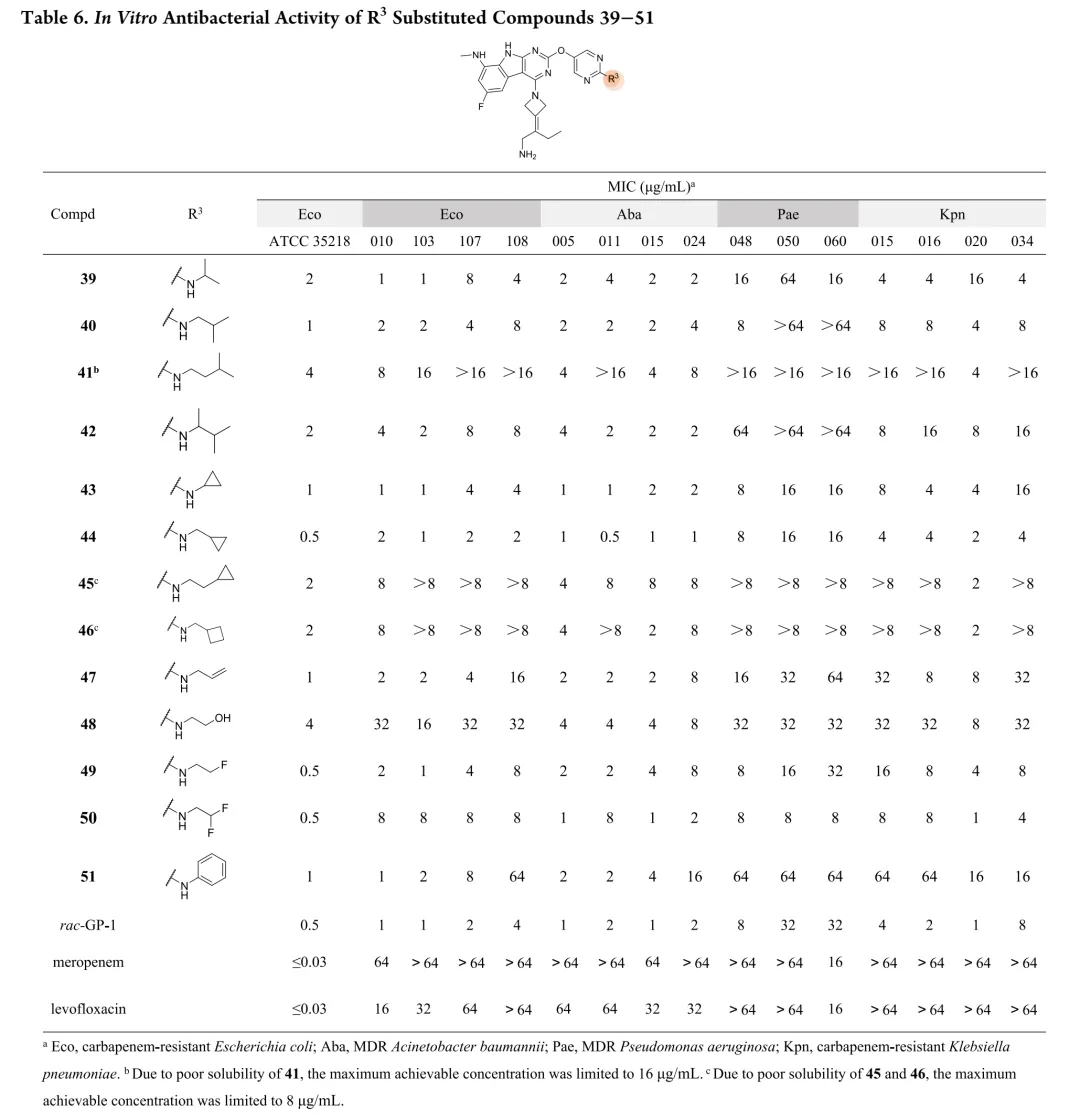
新合成化合物的体外抗菌活性按CLSI指南采用肉汤微量稀释法评估,测试菌株涵盖临床分离的耐多药革兰氏阴性菌,同时以ADP-Glo法测定大肠杆菌GyrB抑制活性。初始优化针对代谢不稳定的C-8位:环化策略将甲胺基团纳入吡唑环得化合物12和13,虽有酶抑制活性但无抗菌活性;在吡咯并[2,3-d]嘧啶骨架引入酰胺基团得化合物14,却导致酶活性降低12倍且丧失抗菌活性。骨架杂化策略将吡啶并[2,3-b]吲哚与吡啶并[4,5-b]吲哚结合,所得化合物15-18活性显著减弱,化合物8与15仅因CH替换N即导致抗菌活性降低8-32倍。C-2位引入芳香杂环以形成与Arg136直接氢键的策略中,化合物20酶抑制活性优于rac-GP-1,但未转化为抗菌效力提升,类似物20-24均丧失活性。鉴于上述策略有限,优化转向嘧啶环R3位点;采用CADD策略通过Grow Scaffold模块生成5382个分子,经两轮分子对接筛选保留LibDock评分≥165.80且结合能≤-40.30 kcal/mol的分子,前10名中9个被合成,多数显示良好活性。结果显示酶抑制与抗菌活性总体相关,但存在例外,故以MIC为构效关系首要指标。筛选表明连接位点含氮杂原子的分子占主导,据此系统考察连接原子与链长、氮原子性质、链分支及末端修饰四方面。
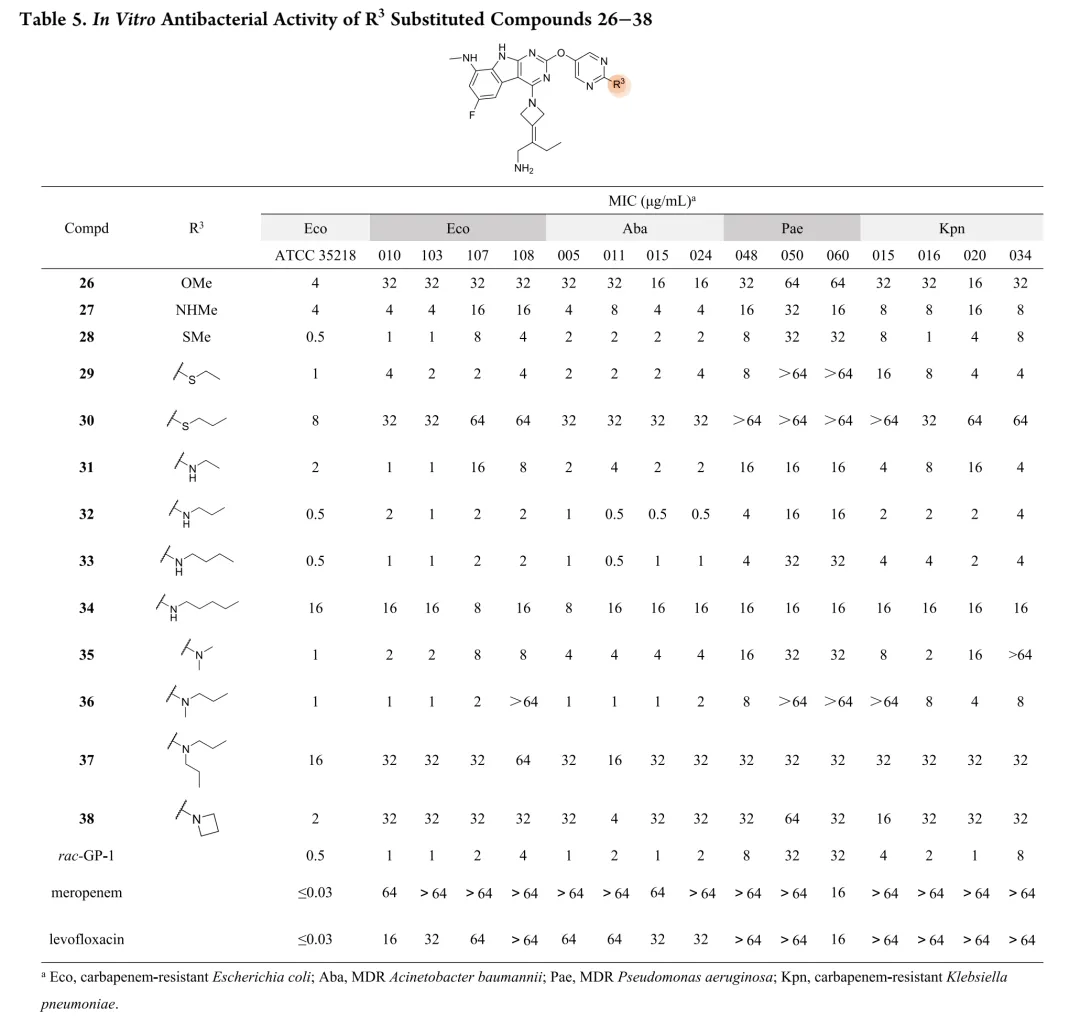
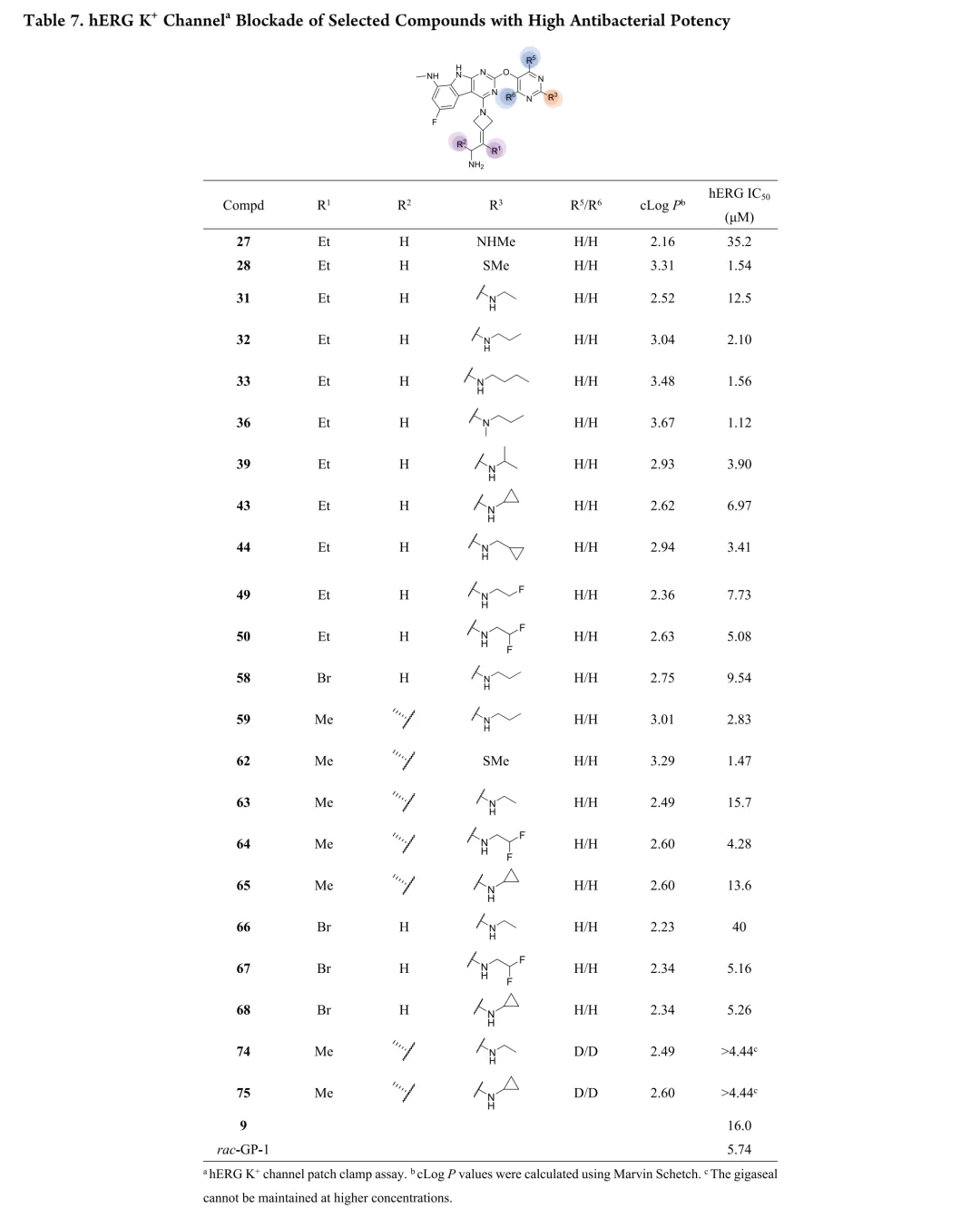
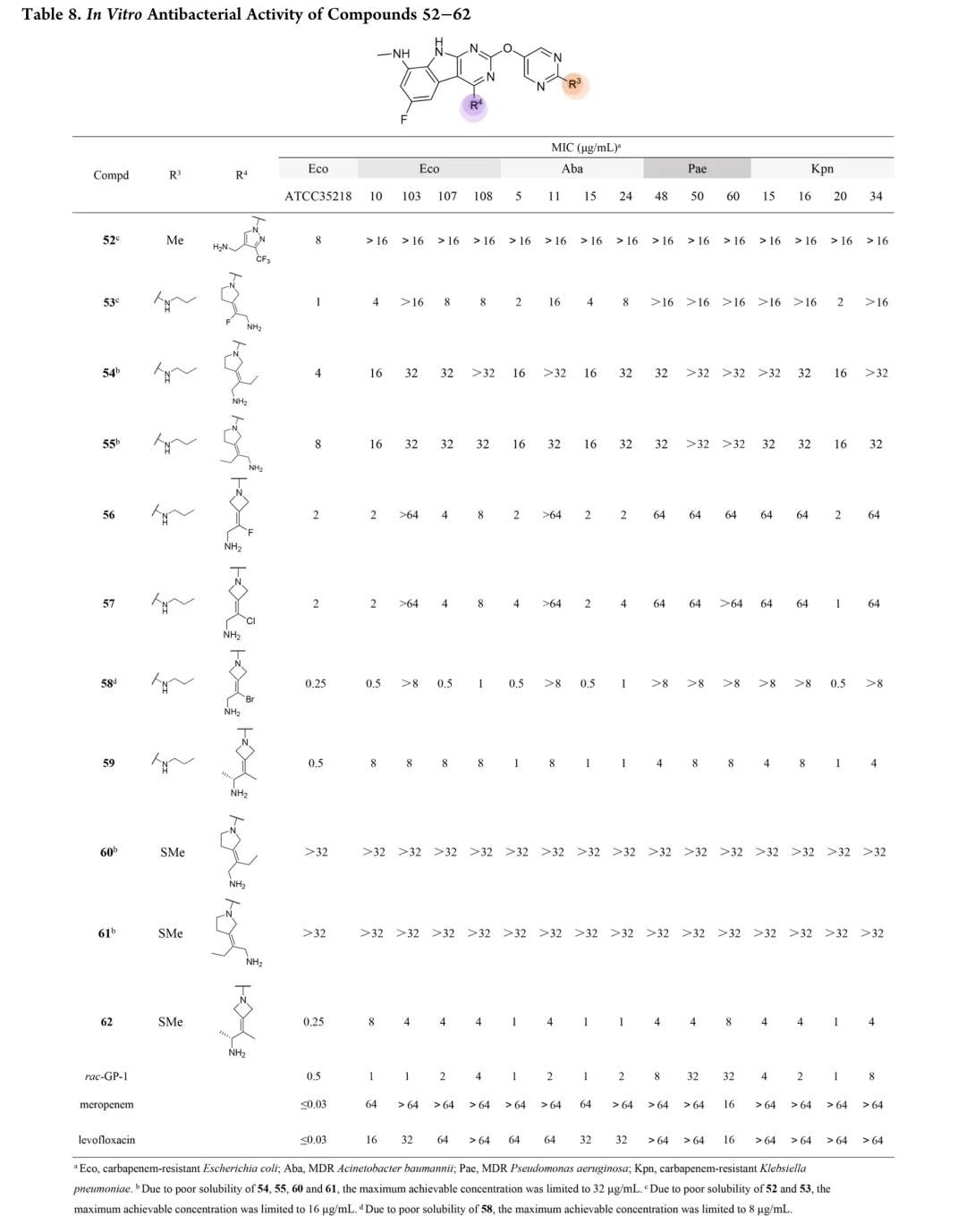
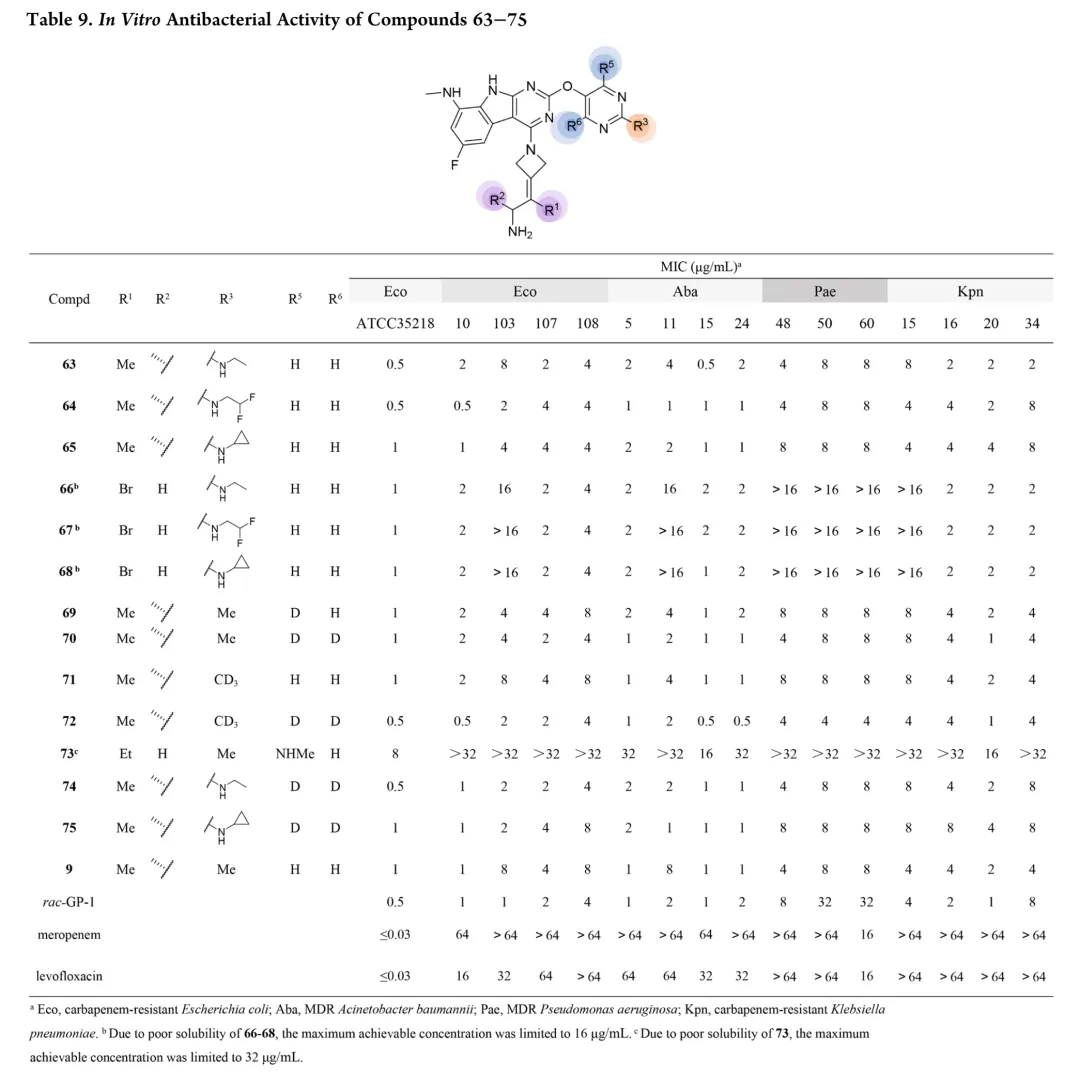
首先考察R3位杂原子影响:甲氧基(26)活性有限,甲硫基(28)广谱活性与rac-GP-1相当,甲氨基(27)对铜绿假单胞菌保持活性但对其他菌株减弱,效力顺序为甲硫基 > 甲氨基 > 甲氧基。链长变化研究显示:烷硫基系列随链延长活性递减,乙硫基(29)活性降低,正丙硫基完全失活;烷氨基则呈抛物线构效关系,2-4碳链(31-33)维持广谱活性,3碳(32)达峰值。胺功能基考察表明:叔胺(35-37)和脂环胺(38)活性降低,最优活性需满足仲胺及2-4碳线性烷基链。支链异构体(39-42)中,异丙氨基(39)活性适度提升,但异丁基(40)和异戊基(41)及α位甲基取代(42)均导致活性下降。末端环化修饰中,环丙基甲基氨基(44)为最强效类似物,而直接环丙基(43)、环丁基(46)及链延长(45)均不利。末端功能基修饰显示:烯烃(47)、羟基(48)、单氟(49)均降低活性,二氟乙氨基(50)对铜绿假单胞菌活性最优但大肠杆菌活性下降,苯氨基(51)对后两者完全失活。hERG评价揭示与cLog P的负相关(R²=0.695):28、32、33、44显示显著抑制(IC50<4μM),31和43风险较低。R4位修饰中,吡唑(52)活性差,吡咯烷(53)可行但乙基替换不利;卤素替换策略中,溴代(58)广谱活性优于rac-GP-1且hERG风险降低(9.54μM),9A衍生物59和62对铜绿假单胞菌活性提升2-8倍且hERG影响最小。综合优化得63和65:广谱活性优于rac-GP-1,hERG IC50>10μM,65的GyrB IC50(12.60nM)更优。氘代研究(69-75)未改善性质,故选定63和65进行药代动力学评价。
④选定类似物的药代动力学研究
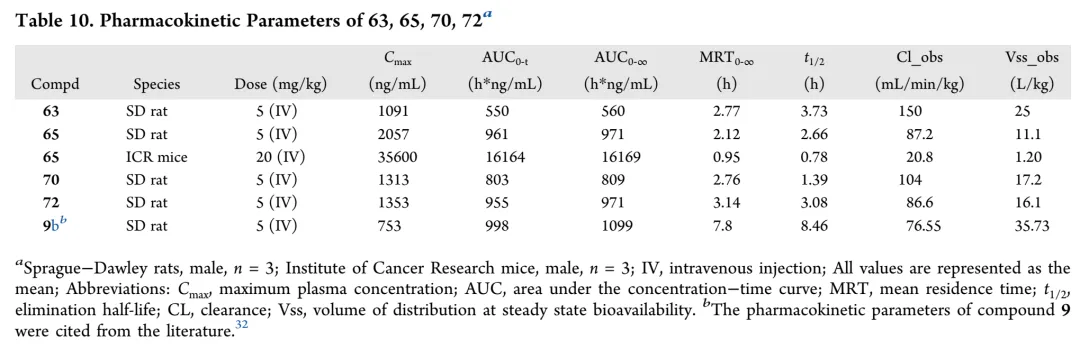
化合物63和65在SD大鼠中单次静脉给药(5 mg/kg)后的药代动力学评价显示:与前体化合物9相比,63的Cmax提高1.45倍,但AUC0-24仅为9的55%,归因于其高血浆清除率(150 mL/min/kg),该加速清除可能源于无位阻乙氨基侧链的细胞色素P450介导氧化;而65采用环丙基氨基取代以空间位阻阻断氧化代谢,显示增强的代谢稳定性,AUC0-24与9相当,Cmax提高2.73倍(2057 ng/mL)。进一步在ICR小鼠中评价65(20 mg/kg,静脉给药)确认其优化特征:Cmax达35600 ng/mL,AUC0-24为16164 h·ng/mL。对于浓度依赖性抗菌药(如氟喹诺酮类),杀菌疗效与Cmax/MIC和AUC0-24/MIC(主要PK/PD指数)的相关性强于半衰期,因此65作为代谢优化类似物,预测其体内疗效优于9。
⑤化合物65的体内疗效评价
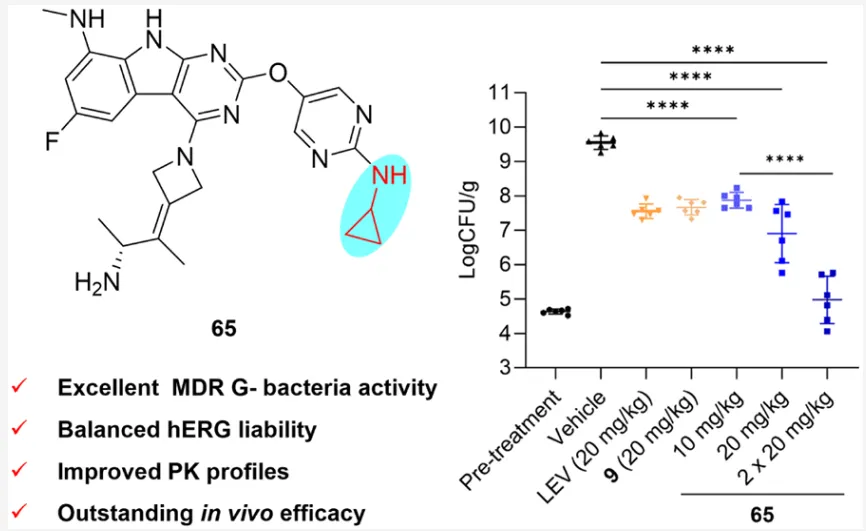
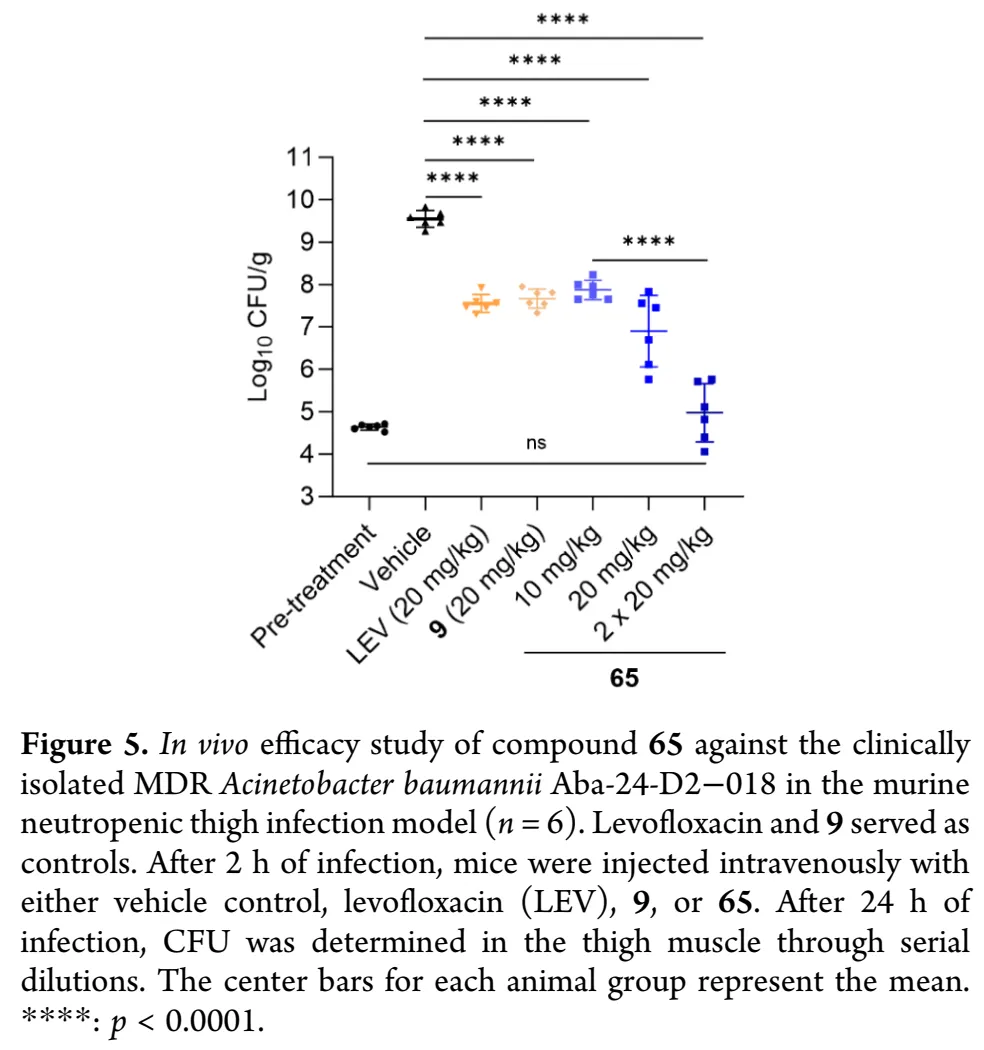
基于有前景的体外活性和优化药代动力学性质,65在中性粒细胞减少小鼠大腿感染模型中评价体内疗效,选用临床分离的耐美罗培南MDR鲍曼不动杆菌Aba-2024-D2-018(MIC:65为0.5 μg/mL,rac-GP-1为1 μg/mL,美罗培南和左氧氟沙均为32 μg/mL),以左氧氟沙星和前体化合物9为阳性对照,静脉注射给药,24小时后通过大腿细菌负荷对数减少量(log10 CFU/g)定量疗效。结果显示,两个阳性对照(左氧氟沙星和9)在20 mg/kg单剂量下产生≤2 log CFU的中等程度降低;65在半数剂量(10 mg/kg)即达到与阳性对照相当疗效,剂量提升至20 mg/kg时产生显著增强的2.65 log CFU降低;值得注意的是,65以20 mg/kg每日两次给药产生深达的4.57 log CFU降低(p<0.0001),相当于>99.99%细菌清除,使细菌负荷降至预处理水平。这些数据证实了65对抗MDR鲍曼不动杆菌感染的优异体内疗效和剂量依赖性。
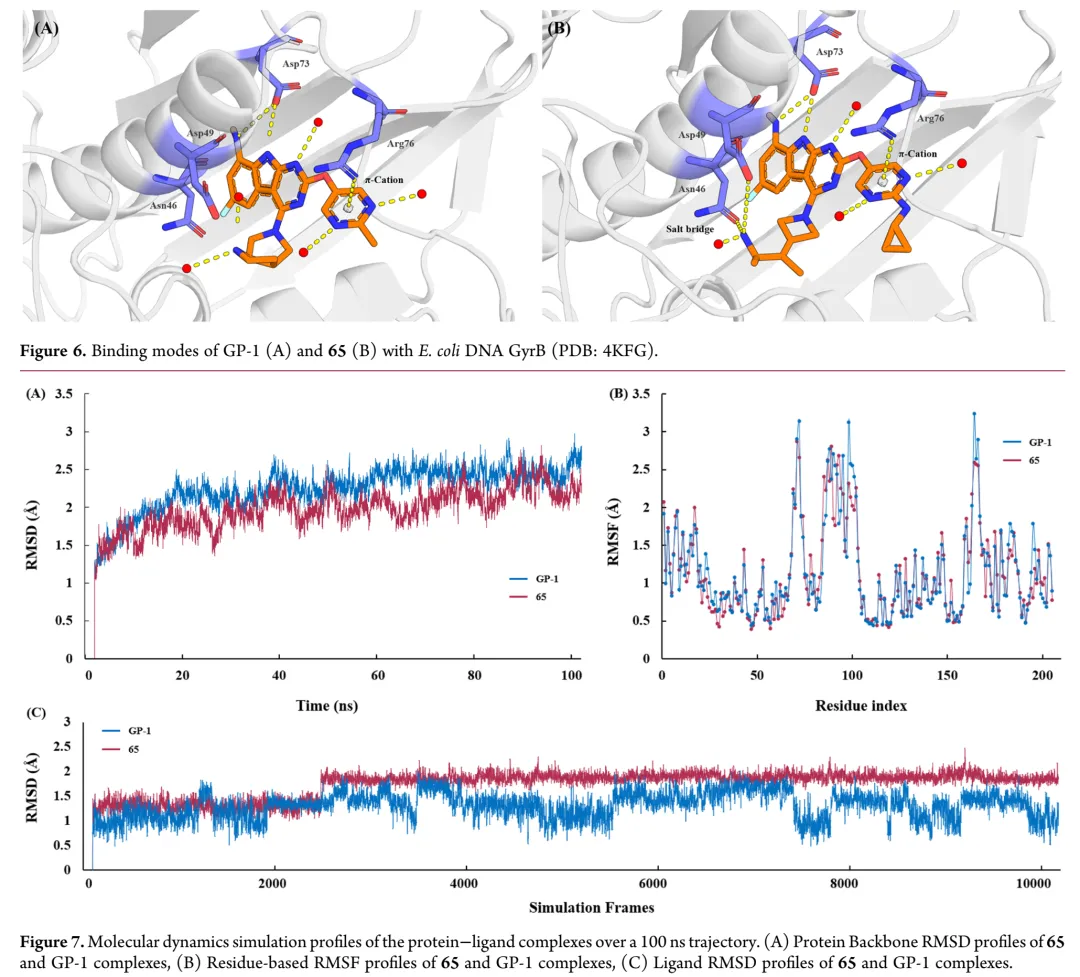
⑥化合物65与大肠杆菌DNA旋转酶B亚基的结合模式
为探究65和GP-1抑制活性的结构基础,首先通过分子对接预测两者在大肠杆菌DNA旋转酶B亚基ATP结合域(PDB: 4KFG)中的结合取向:两配体共享保守骨架,通过常规氢键网络(与Asp73)、四个保守水介导氢键及与Arg76的π-阳离子相互作用锚定于结合口袋。尽管相似,65更契合口袋并形成额外稳定相互作用,包括与Asp49的额外盐桥和与Asn46的新氢键;相互作用环图显示65具有更优的口袋占据。值得注意的是,R3基团虽预期处于溶剂暴露区,但65的环丙基氨基实际埋藏于由周围残基(特别是Phe100)形成的亚口袋中,采取独特弯曲构象,促进关键疏水接触,与疏水取代基在R3位增强活性的构效关系趋势一致。为评估动态条件下的复合物稳定性,进行100 ns分子动力学模拟:尽管两化合物实验IC50值相近,但动态行为显示差异——65复合物的主链RMSD更低且更稳定(~1.98 Å vs ~2.25 Å),表明契合更紧密、全局构象偏离更小;RMSF分析显示高度重叠模式,表明结合未扰动整体蛋白结构,活性口袋关键残基(Asn46、Asp49、Asp73、Arg76)保持低RMSF值(<1.0 Å),环区在65复合物中波动降低,提示局部稳定性增强。配体RMSD曲线证实两化合物平衡后均达稳定平台且无解离:GP-1保持在0.8-1.5 Å窄范围,65显示略高但稳定的偏离(1.6-2.0 Å),反映优化契合所需的细微构象适应性调整,最终产生更优几何互补性,体现为更稳定的蛋白主链(~1.98 Å vs 2.25 Å)和显著改善的总结合自由能(-39.73 vs -29.22 kcal/mol)。分子对接结果提示65保持与GP-1一致的结合模式同时可能形成额外稳定相互作用,100 ns分子动力学模拟为对接预测的结合模式提供动态支持,显示65复合物在模拟时间框架内的结构稳定性趋势。
近年来,靶向DNA旋转酶非喹诺酮结合位点的ATP酶抑制剂成为抗感染研究热点,吡啶并[4,5-b]吲哚衍生物虽具广谱抗耐多药革兰氏阴性菌活性,但受限于体外活性不足、hERG抑制、药代动力学缺陷及有限体内疗效。本研究设计合成系列新类似物:C-8位修饰及骨架杂化策略未成功;经CADD指导系统优化R3位点,发现氮杂原子最优,烷氨基呈链长抛物线构效关系,2-4碳链维持广谱活性,环丙基末端(44)活性最佳;首次揭示hERG抑制与cLog P负相关(R²=0.695)。整合构效关系与构效毒性关系得化合物65:GyrB IC50 12.6 nM,hERG IC50>10μM,大鼠Cmax较化合物9提高2.73倍,小鼠静脉给药20 mg/kg后Cmax 35600 ng/mL、AUC0-24 16164 h·ng/mL;在耐多药鲍曼不动杆菌感染模型中,20 mg/kg每日两次给药降低细菌负荷4.57 log10 CFU/g,实现>99.99%清除,为首个体内疗效超越氟喹诺酮类的该骨架衍生物,值得进一步开发。
文献详细全面信息请跳转原文阅读:
https://doi.org/10.1021/acs.jmedchem.6c00278
欢迎合理转载,请写明来源于【PharmaView】,公众号以个人学习和分享最新科研成果为目的,本文内容为阅读文献提炼总结的内容,因学识有限,难免有所疏漏和谬误,恳请批评指正。标记【原创】指原创编译,是出于便于传播目的,著作权归出版社与作者所有。







 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
