1. 定义并验证高可塑性细胞状态(HPCS)
以Slc4a11为特异性标志物,构建基因工程小鼠模型,证实HPCS是肺癌细胞状态转换的核心枢纽,兼具双向分化潜能。
2. 明确HPCS的关键病理功能
HPCS驱动早期肿瘤恶变、维持肿瘤异质性,其消融可阻断良性-恶性转化,显著抑制已建立肿瘤的生长与负荷。
3. 揭示HPCS介导治疗耐药机制
化疗与KRAS靶向治疗后,HPCS存活并分化为耐药细胞群,联合消融HPCS可实现肿瘤近完全清除,逆转治疗抵抗。
4. 证实HPCS的泛癌与再生相关性
HPCS样状态普遍存在于多种癌种及损伤修复的上皮组织中,靶向uPAR的CAR-T细胞可有效清除HPCS,具广谱治疗潜力。
肺癌细胞可塑性是肿瘤进展与治疗耐药的关键驱动因素,现有放化疗、靶向治疗难以阻断细胞状态转换,晚期患者预后较差。高可塑性细胞状态(HPCS)被证实是细胞状态转换的核心枢纽,但针对该状态的靶向策略尚不成熟。研究旨在明确HPCS在肺癌进展中的功能,评估靶向HPCS的治疗效果,并揭示其调控肿瘤异质性与耐药的分子机制。
如果你也想利用GBD、机器学习、孟德尔随机化、CHARLS/NHANES等数据库进行生信分析,欢迎关注并后台咨询!
可塑性——即细胞发生表型转变的能力——驱动癌症进展和治疗抵抗。最近的研究表明,实体肿瘤中的可塑性集中于一小部分癌细胞,但针对这种高可塑性细胞状态(HPCS)的体内原位功能研究尚属缺乏。
研究构建KP肺癌小鼠模型,通过Slc4a11位点敲入报告基因标记高可塑性细胞状态(HPCS);结合谱系追踪、分泌型荧光素酶纵向监测及细胞消融技术,探究HPCS功能;利用单细胞测序解析其细胞状态转换;通过化疗、靶向治疗结合消融实验验证治疗敏感性;借助uPAR CAR-T细胞靶向HPCS,同时跨癌种及再生上皮数据集验证HPCS保守性。
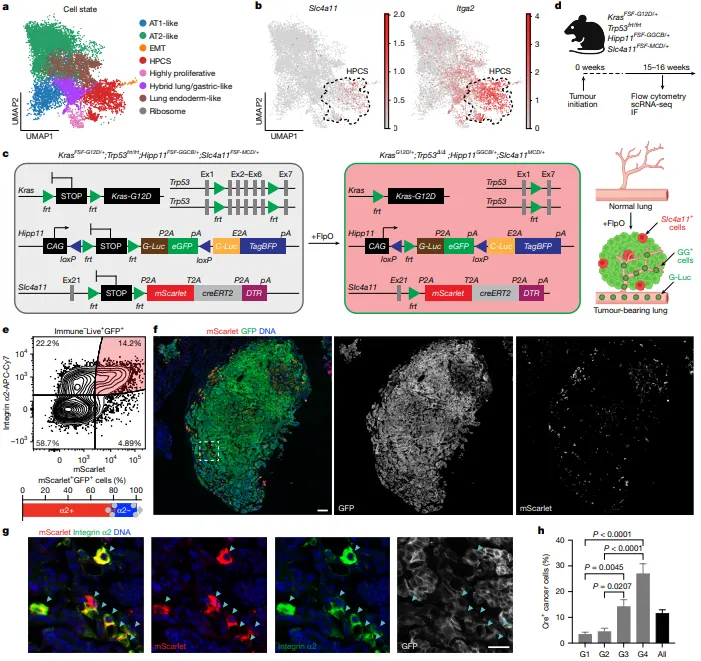
单细胞转录组测序识别出LUAD的8种细胞状态,确定Slc4a11为HPCS特异性标记基因、Itga2为辅助标记;构建Slc4a11-MCD报告系统可精准标记HPCS,流式显示17.0±4.3% GFP+癌细胞为mScarlet+ HPCS,78.4±1.7%共表达Integrin α2;免疫荧光证实HPCS呈小簇/单细胞分布,且占比随肿瘤分级升高而显著增加(图1)。
图1.Slc4a11MCD/+报告系统可在体内标记高可塑性细胞状态(HPCS)
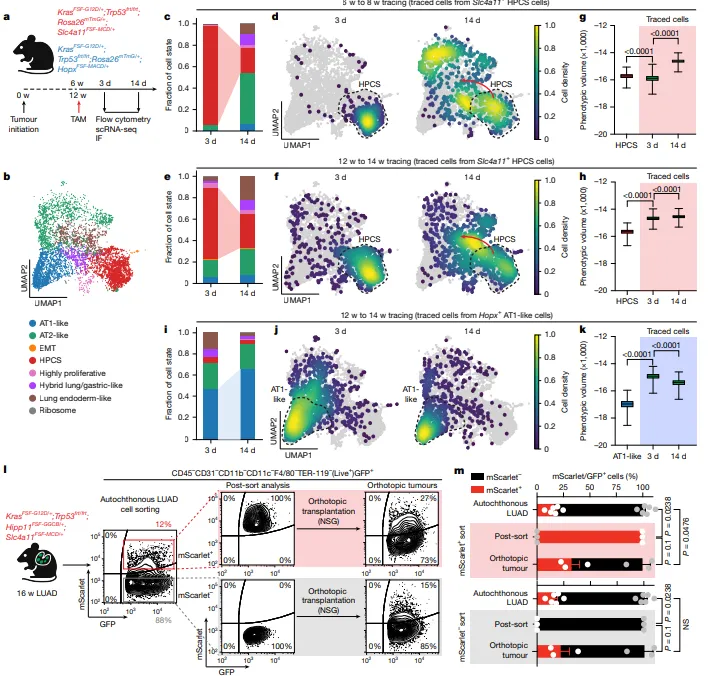
通过谱系追踪证实,HPCS具有强大的细胞状态转换能力,可分化为腺瘤和腺癌中所有观察到的癌细胞状态(图2c-f),且追踪14天后表型体积显著增加,是肿瘤异质性的核心驱动因素(图2g-h)。
对比AT1样细胞的谱系追踪显示,AT1样细胞可塑性低,仅能维持自身状态且表型体积收缩(图2i-k)。
移植实验表明,非HPCS细胞可动态获得HPCS表型,且HPCS形成肿瘤的效率是non-HPCS细胞的9倍(图2l-m)。
图2.高可塑性细胞状态(HPCS)是体内细胞状态转换的核心枢纽
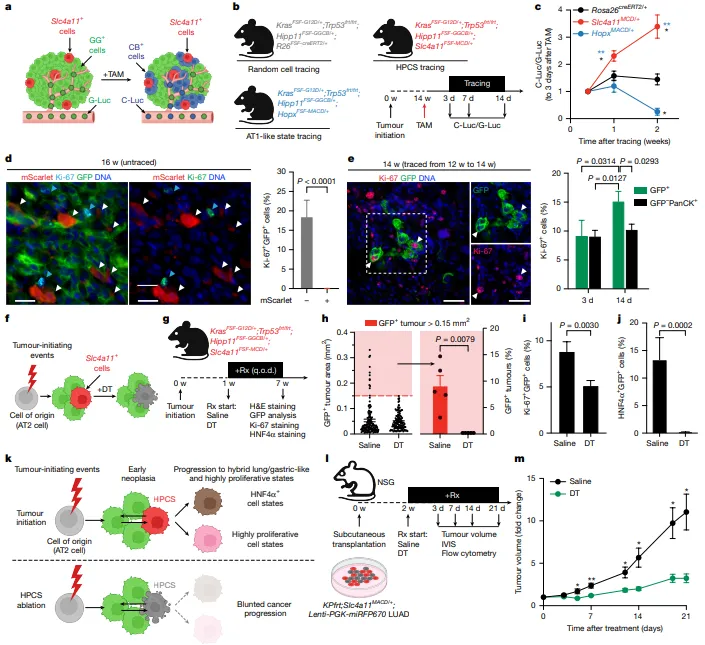
通过分泌型荧光素酶纵向追踪发现,HPCS来源细胞的增殖能力显著高于随机标记细胞和AT1样细胞(图3c)。
HPCS本身呈静息状态(Ki-67阴性),但其分化后代快速进入细胞周期并驱动克隆扩张(图3d-e)。
早期消融HPCS可阻断不典型腺瘤样增生向腺瘤进展,减少大肿瘤病灶和高增殖细胞比例(图3f-i),而晚期消融则显著降低肿瘤负荷(图3m),证实HPCS对肿瘤进展和维持至关重要。
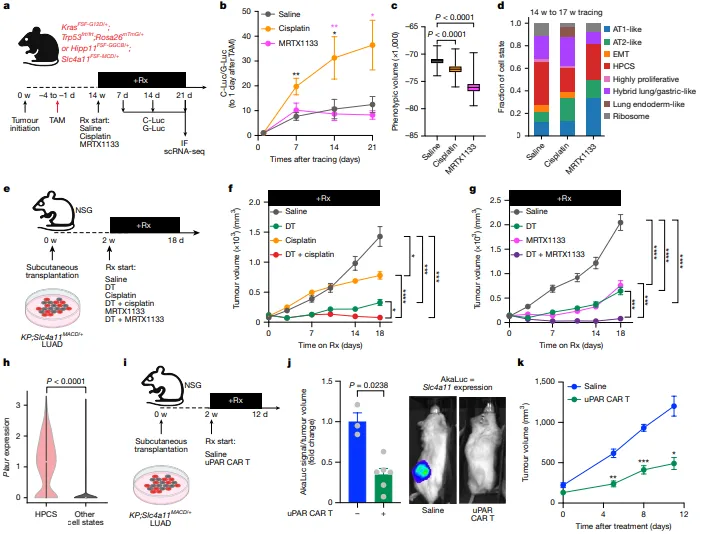
顺铂与MRTX1133主要清除non-HPCS,HPCS存活并富集于MRD;scRNA-seq证实HPCS可分化为多种耐药细胞状态。HPCS消融与化疗/靶向治疗联合可实现近完全肿瘤根除;此外,HPCS高表达Plaur,uPAR CAR-T可有效消融HPCS并显著抑瘤(图4)。
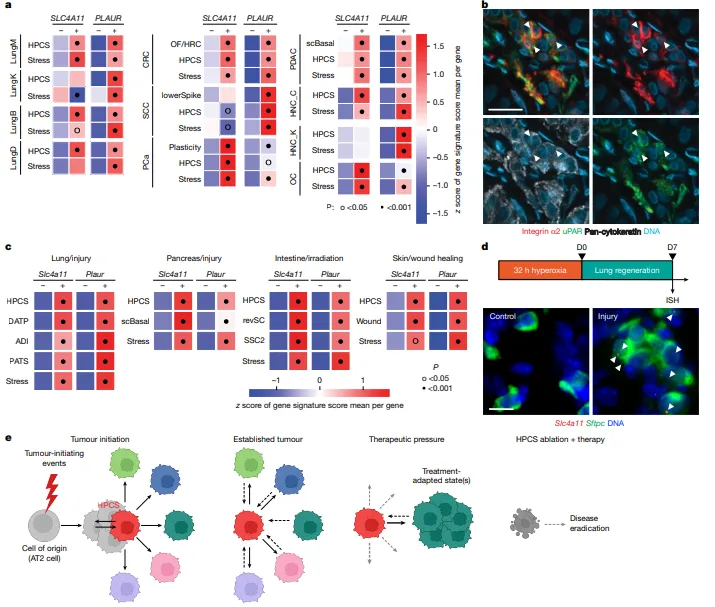
HPCS特征在结直肠癌、胰腺癌、前列腺癌等多种癌种中普遍存在,且与耐药、复发相关的细胞状态高度相关(图5a)。
人类LUAD组织中存在uPAR+Integrin α2+的HPCS样细胞(图5b)。
HPCS签名、Slc4a11和Plaur在肺、胰腺、肠道、皮肤的损伤再生上皮中显著富集(图5c),且高氧损伤后小鼠肺AT2细胞中Slc4a11表达诱导上调(图5d),表明HPCS是癌细胞劫持上皮再生程序形成的可塑性状态。
图5.一种类似HPCS的状态,其特征为Slc4a11和Plaur的存在,在多种癌变组织和再生上皮中普遍存在
研究确立了HPCS作为癌细胞状态间相互转变的关键枢纽。靶向肺癌及其他癌瘤中的HPCS可能抑制癌症进展并根除治疗抵抗。