农业农村部沼科所王文国/张宏伟&西华大学李光照,最新Angew!非晶相调控TiO₂-ZrO₂界面连接以调控自由基路径助力高选择性光催化甲烷氧化
非晶态氧化物具有独特的短程有序性和配位灵活性,但其在光催化甲烷转化为液体产物过程中调控界面自由基化学并抑制过氧化的能力,仍然是一项关键的前沿问题。
2026年04月14日,农业农村部成都沼气科学研究所王文国/张宏伟、西华大学李光照团队合作在Angewandte Chemie International Edition期刊发表题为“Amorphous-Phase Tailoring of TiO2–ZrO2 Interfacial Linkages Regulates Radical Pathways for Highly Selective Photocatalytic Methane Oxidation”的研究论文,农业农村部成都沼气科学研究所/西华大学周圣荣为论文第一作者,张宏伟、李光照、王文国、天津大学宋辉为论文共同通讯作者。
第一作者:周圣荣
通讯作者:张宏伟、李光照、王文国、宋辉
通讯单位:农业农村部成都沼气科学研究所、西华大学、天津大学
论文DOI:10.1002/anie.9660324
该研究策略性地构建了TiO₂-ZrO₂结构,其中ZrO₂相从非晶态(TiO₂-ZrO₂-A)精确调控至晶态(TiO₂-ZrO₂-A800),以阐明结构无序对有氧CH₄功能化的影响规律。多模态表征,包括X射线吸收近边结构(XANES)、扩展X射线吸收精细结构(EXAFS)、AC-HRTEM、瞬态电子顺磁共振(EPR)、原位漫反射红外傅里叶变换光谱(DRIFTS)以及密度泛函理论DFT计算,揭示了非晶ZrO₂相相较于其晶态对应物富集了表面氧空位,并建立了稳健的Ti-O-Zr界面连接。这种构型增强了电荷分离,并支持更高的有效稳态空穴可用性以实现高效C-H键活化,同时有效调节了源自水的活性氧物种的通量。在室温下,TiO₂-ZrO₂-A实现了高达98.2%的优异液体含氧产物选择性,其显著优于其晶态类似物,并能抵抗深度氧化为CO₂。动力学分析揭示了*CH₃和*CH₃O中间体的快速形成,这与非晶界面更快中间体转化和减少的过氧化现象一致,并且与DFT计算出的有利于甲烷活化而非非选择性活性氧物种(ROS)生成的能垒相符。这些发现证实了非晶相工程是一种调控光催化C-H转化中反应路径和选择性的动力学阀门。
甲烷(CH₄)是天然气和沼气的主要成分,通过将其选择性转化为高价值的液体含氧产物(如甲醇(CH₃OH)和甲醛(HCHO)),作为一种用于可持续循环碳经济的战略性一碳(C1)原料。尽管取得了显著进展,但传统的热催化CH₄转化仍然受到C-H键极端稳定性(约439 kJ/mol)的阻碍,通常需要在高温和/或高压条件下操作(例如,蒸汽重整温度为700°C-1000°C,压力为15-30 bar),导致高能耗、复杂的反应工程以及频繁的过度氧化为CO₂,从而降低了碳效率。这些限制激发了人们对光催化的日益增长的兴趣,该技术可以在室温下通过直接利用光子产生电荷载流子和活性氧物种(ROS)来活化CH₄。
在光催化有氧CH₄氧化中,光生空穴(h⁺)和RROS(特别是羟基自由基(•OH)和氢过氧自由基(•OOH))协同作用,裂解强C-H键并将产物形成导向液态C1含氧产物(CH₃OH、HCHO)。为了提高反应速率和选择性,研究人员采用了负载贵金属助催化剂(Au、Pd、Ru等)的半导体平台(例如TiO₂、ZnO、WO₃),这些助催化剂可以改善光捕获、加速电荷分离,并催化水氧化和氧还原以提供ROS。然而,由于催化剂表面存在异质性的位点集合和多种自由基路径,结构-选择性关系仍然不明确,深度氧化为CO₂常常与目标含氧产物路径竞争。
表面结构对这些路径具有首要影响。晶面依赖的原子排列和配位可以调控吸附能、载流子分离以及ROS的通量,从而调节活性和选择性。然而,大多数研究都集中在结晶良好的晶面上,而非晶相(具有其短程有序性、配位灵活性和丰富的缺陷化学)的催化作用则远未被充分理解。特别是,半导体界面上的非晶氧化物如何调控氧空位(Oᵥ)、界面键合、电荷传输以及含氧中间体的停留时间/转移,尚未得到系统阐明。
在此,该研究通过使用具有非晶ZrO₂的TiO₂复合材料(TiO₂-ZrO₂-A),并将其与晶化的四方晶类似物(TiO₂-ZrO₂-A800)以及物理混合物进行对比,来解决这一研究空白。研究人员发现,非晶ZrO₂富集了Oᵥ并构建了Ti-O-Zr界面连接,这些连接增强了载流子分离,同时调节了ROS通量。在接近25°C的水中、紫外-可见光照射下,TiO₂-ZrO₂-A通过抑制过度氧化为CO₂,实现了高达98.2%的液态含氧产物选择性。机理探针(结合自旋捕获的EPR、原位DRIFTS)与XANES/EXAFS和DFT计算结果表明,TiO₂-ZrO₂-A上具有更高的h⁺可用性和受控•OH/•OOH生成,这有利于*CH₃/*CH₃O中间体,但不利于深度氧化。该研究发现确立了非晶相工程作为一种强大的动力学阀门,用于指导反应路径,为在温和条件下进行C-H键官能团化的高选择性光催化系统设计提供了新视角。
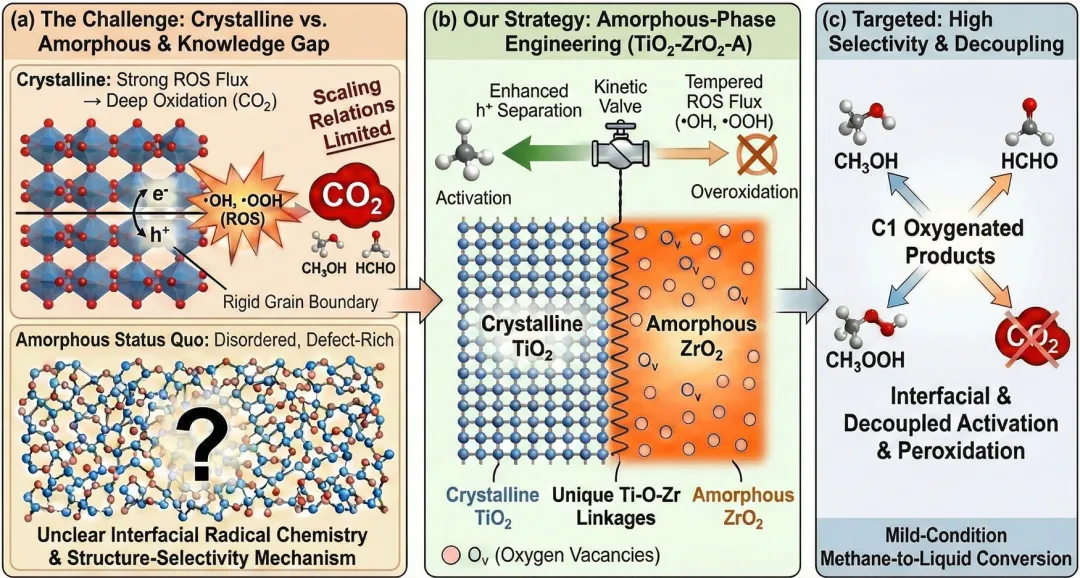
示意图1|用于高选择性光催化甲烷氧化的非晶相工程的概念框架。
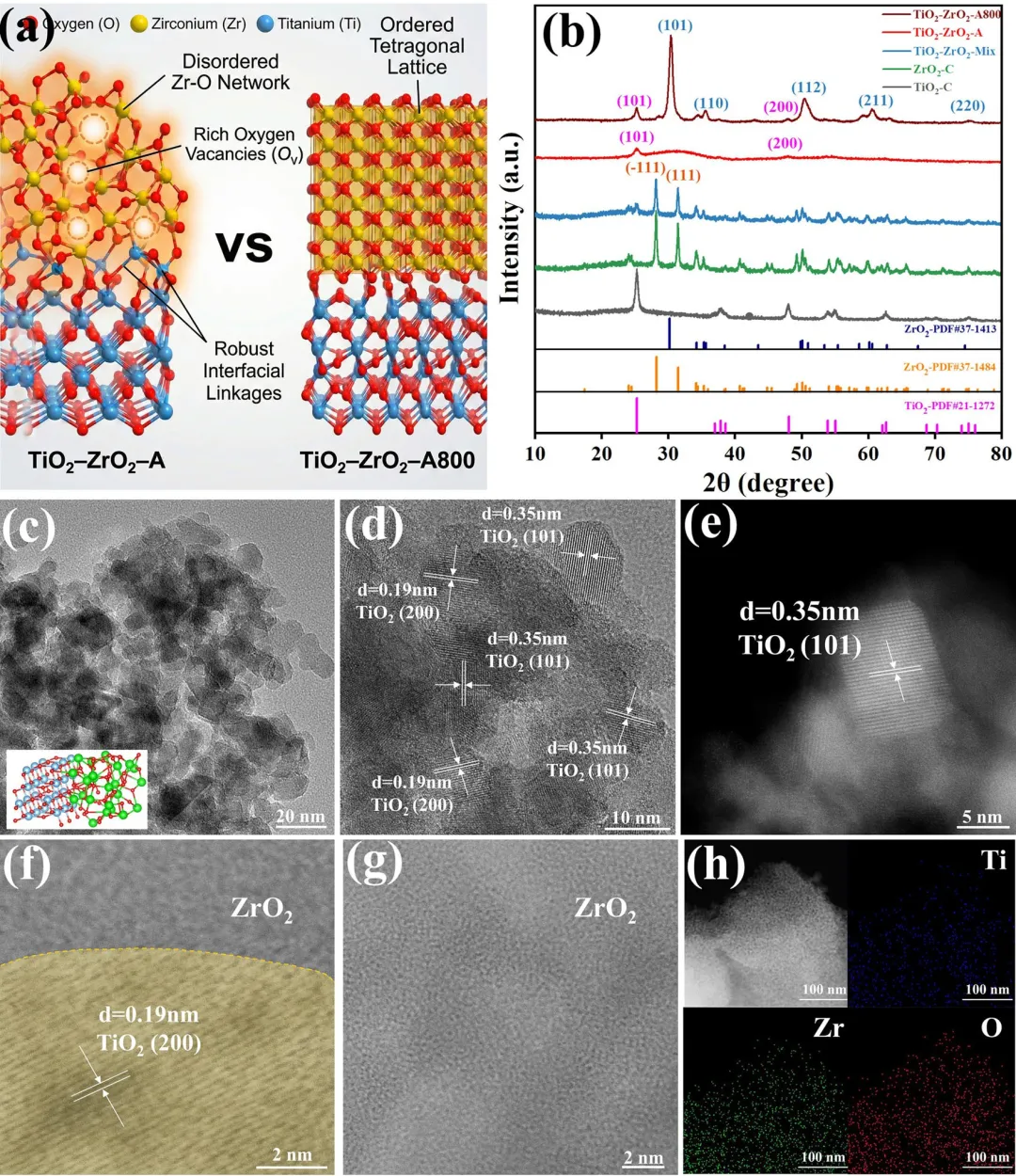
图1|(a) TiO₂-ZrO₂-A和TiO₂-ZrO₂-A800结构示意图;(b) TiO₂-C、ZrO₂-C、TiO₂-ZrO₂-Mix、TiO₂-ZrO₂-A和TiO₂-ZrO₂-A800的XRD图谱。(c, d) TiO₂-ZrO₂-A的TEM图像。(e-g) TiO₂-ZrO₂-A的AC-TEM图像。(h) TiO₂-ZrO₂-A的HAADF图像以及对应的Ti、Zr和O的EDS元素面分布图。
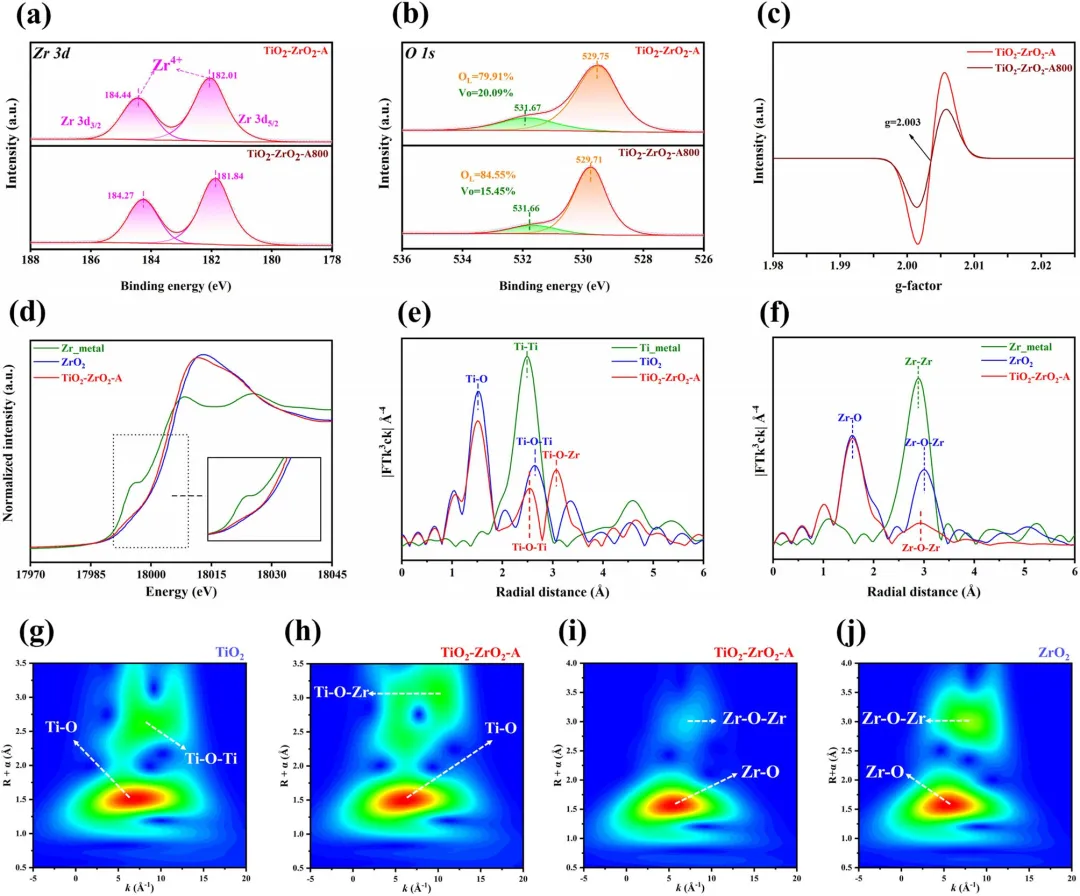
图2|(a, b) TiO₂-ZrO₂-A和TiO₂-ZrO₂-A800的XPS谱图:(a) Zr 3d; (b) O 1s。(c) TiO₂-ZrO₂-A和TiO₂-ZrO₂-A800的EPR谱图。(d) TiO₂-ZrO₂-A的Zr K-edge XANES谱图,以Zr箔和ZrO₂为参照;(e) TiO₂-ZrO₂-A的Ti K-edge EXAFS的k³加权傅里叶变换,以Ti箔和TiO₂为参照。(f) TiO₂-ZrO₂-A的Zr K-edge EXAFS的k³加权傅里叶变换,以Zr箔和ZrO₂为参照。(g, h) (g) TiO₂和(h) TiO₂-ZrO₂-A的Ti K-edge EXAFS的小波变换等高线图。(i, j) (i) TiO₂-ZrO₂-A和(j) ZrO₂的Zr K-edge EXAFS的小波变换等高线图。
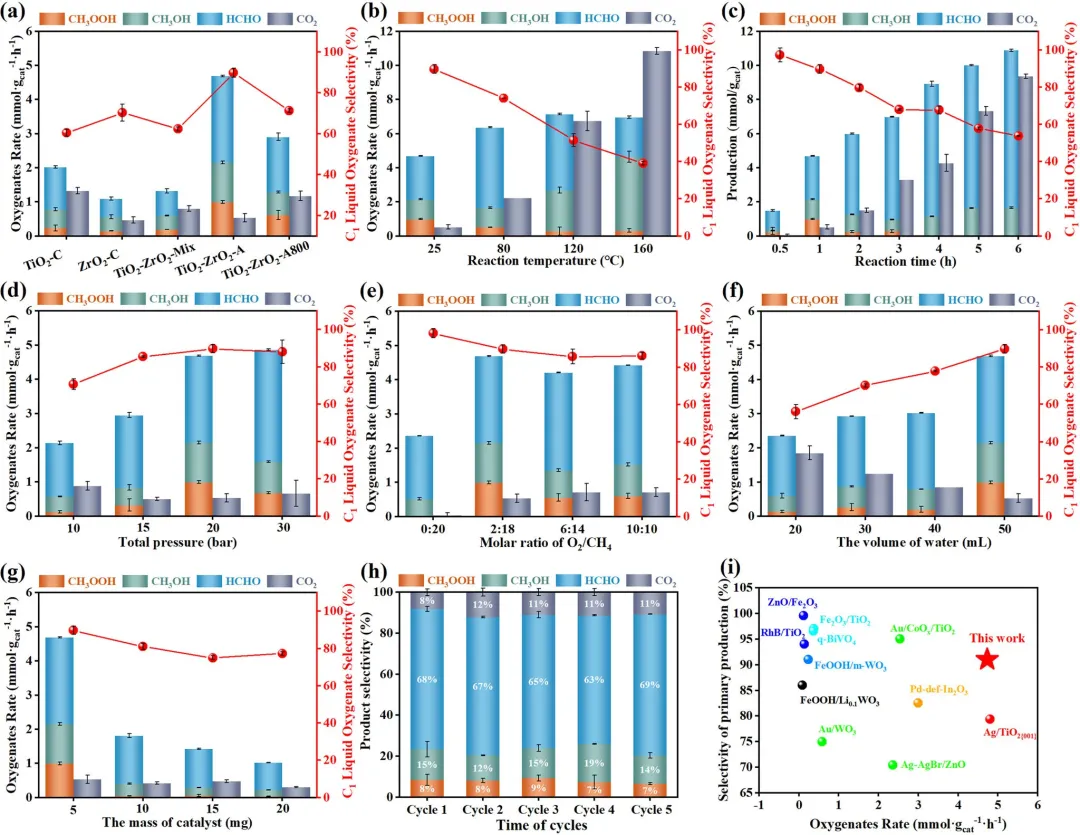
图3|光催化有氧氧化CH₄为C1含氧产物在TiO₂-ZrO₂催化剂上的性能。(a) 在TiO₂-C、ZrO₂-C、TiO₂-ZrO₂-Mix、TiO₂-ZrO₂-A和TiO₂-ZrO₂-A800上的产物分布和C1-含氧产物选择性。TiO₂-ZrO₂-A的反应参数优化:(b) 反应温度,(c) 反应时间,(d) 总压,(e) O₂/CH₄摩尔比,(f) 水体积,以及(g) 催化剂质量。(h) TiO₂-ZrO₂-A在五次连续循环实验中的循环稳定性。(i) 与代表性文献光催化剂的含氧产物产率和主要产物选择性对比。标准反应条件:5 mg催化剂,50 mL H₂O,25°C,2 bar O₂,18 bar CH₄,1小时,Xe灯(200-2500 nm),辐照度为1900 mW cm⁻²。
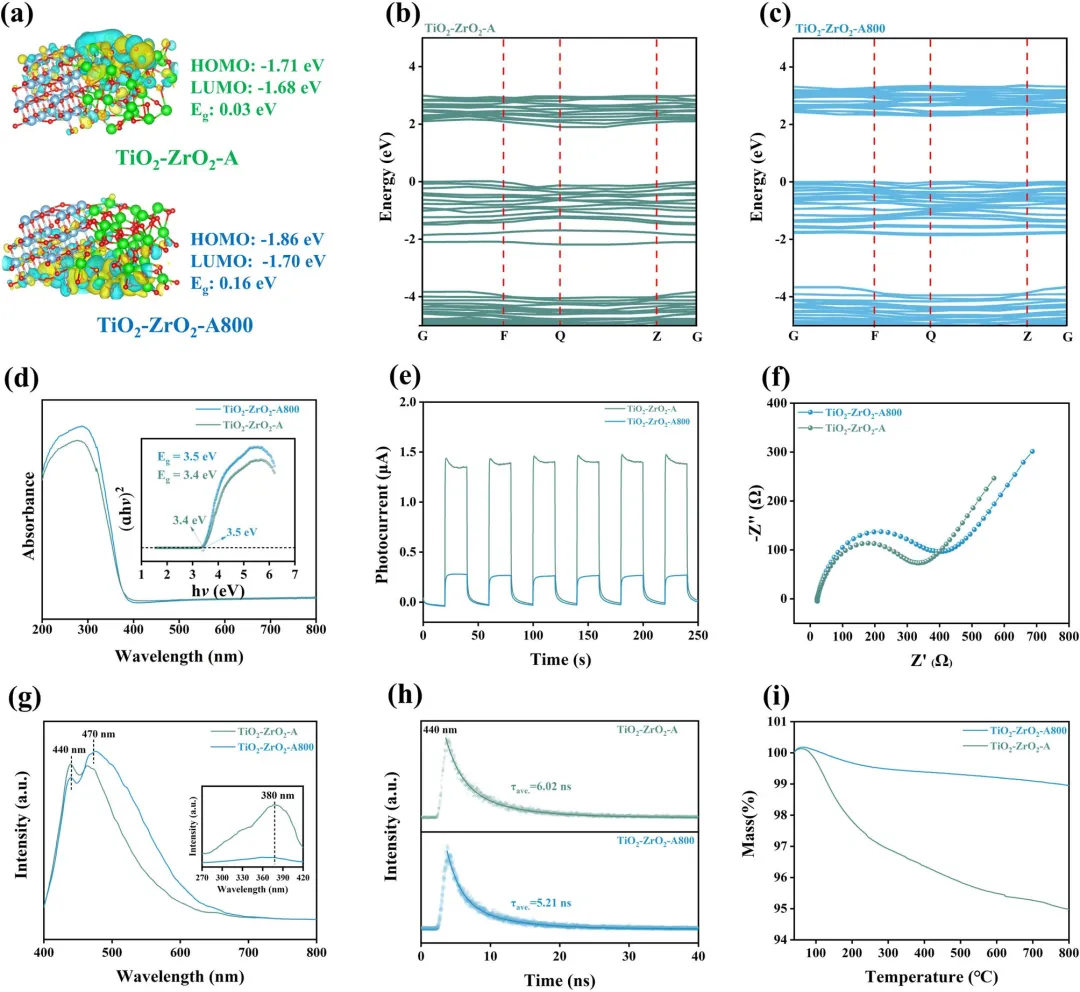
图4|(a) DFT计算的TiO₂-ZrO₂-A和TiO₂-ZrO₂-A800的HOMO和LUMO分布。(b, c) DFT计算的(b) TiO₂-ZrO₂-A和(c) TiO₂-ZrO₂-A800的能带结构。(d) UV-Vis漫反射光谱以及用于估算光学带隙的Tauc图。(e) 在斩波光照下的瞬态光电流响应。(f) EIS Nyquist图。(g) 稳态PL光谱。(h) TRPL衰减曲线。(i) TG曲线。
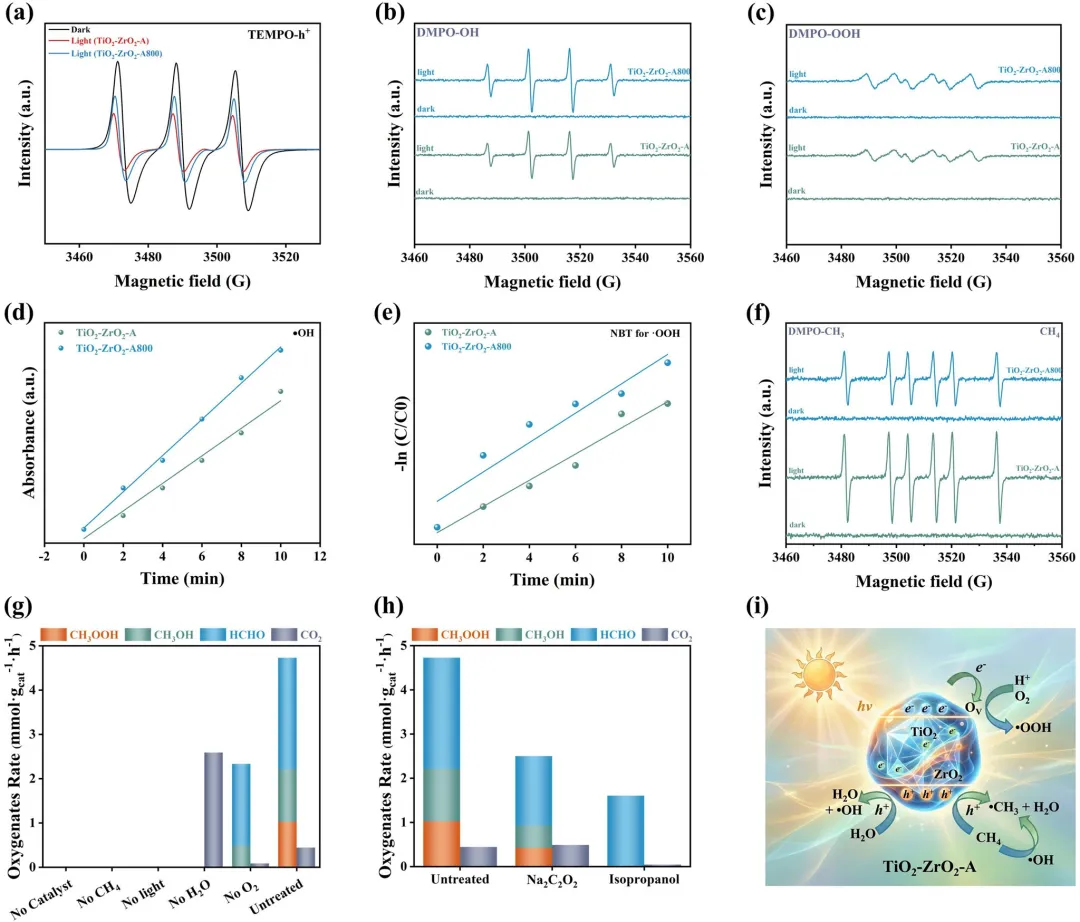
图5|(a) 在暗处和光照条件下,用于探测TiO₂-ZrO₂-A和TiO₂-ZrO₂-A800中光生h⁺的TEMPO-h⁺加合物的EPR谱图。(b, c) 在空气饱和的CH₃OH中,辐照下用于检测(b) 羟基自由基(•OH, DMPO–OH)和(c) 氢过氧自由基(•OOH, DMPO–OOH)的TiO₂-ZrO₂-A和TiO₂-ZrO₂-A800的自旋捕获EPR谱图。(d) 通过乙酰丙酮比色法对•OH进行定量。(e) 通过硝基四氮唑蓝光降解动力学对•OOH进行定量。(f) 辐照下在TiO₂-ZrO₂-A和TiO₂-ZrO₂-A800上生成的甲基自由基(•CH₃, DMPO–CH₃)的自旋捕获EPR谱图;对于DMPO–•CH₃检测,催化剂悬浮液预先用CH₄饱和并加入异丙醇以淬灭•OH干扰。(g) TiO₂-ZrO₂-A在不同反应条件下的对照实验。(h) 在存在Na₂C₂O₄(空穴捕获剂)或异丙醇(•OH捕获剂)时TiO₂-ZrO₂-A的淬灭实验。(g, h)的反应条件:5 mg TiO₂-ZrO₂-A,50 mL溶液,18 bar CH₄,2 bar O₂,25°C,辐照1小时。(i) 在TiO₂-ZrO₂-A上进行光催化CH₄氧化的反应路径示意图。
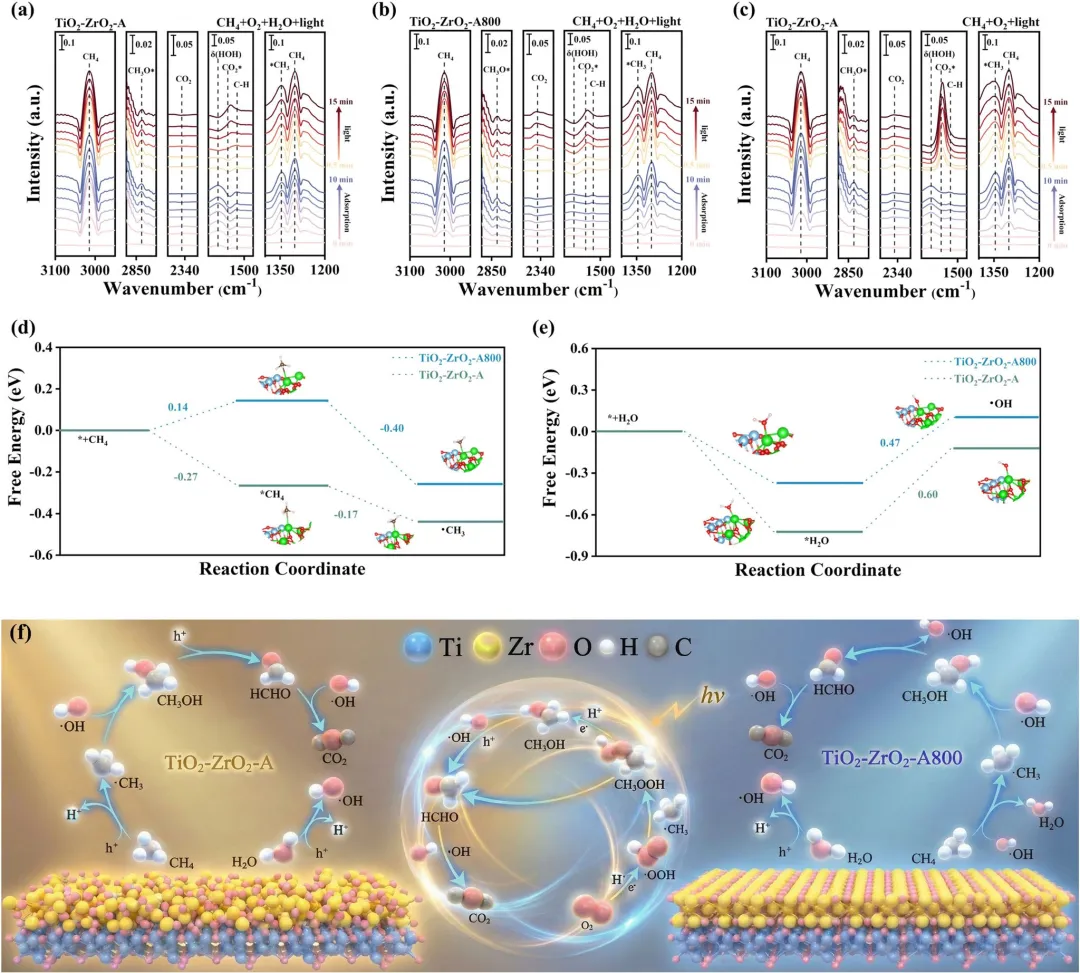
图6|(a, b) 在引入CH₄/O₂/H₂O混合物后,在(a) TiO₂–ZrO₂–A和(b) TiO₂–ZrO₂–A800上记录的原位DRIFTS谱图。(c) 在无水条件下,TiO₂–ZrO₂–A样品的原位DRIFTS谱图。(d) DFT计算的CH₄在TiO₂–ZrO₂–A和TiO₂–ZrO₂–A800上活化的吉布斯自由能图。(e) DFT计算的H₂O在TiO₂–ZrO₂–A和TiO₂–ZrO₂–A800上解离的吉布斯自由能图。(f) 在TiO₂–ZrO₂催化剂上选择性氧化CH₄为C1液态含氧产物的光催化机理示意图,重点突出了TiO₂–ZrO₂–A与TiO₂–ZrO₂–A800上不同的自由基路径和过氧化途径。
总之,该研究成功开发了一系列TiO₂–ZrO₂复合光催化剂,并证实非晶相工程是一种用于引导CH₄选择性氧化为液态含氧产物的有效策略。该研究结果揭示,TiO₂–ZrO₂–A催化剂中的非晶ZrO₂相在抑制CH₄过氧化为CO₂(传统光催化系统中的常见缺陷)方面发挥着决定性作用。在优化的近环境条件下,TiO₂–ZrO₂–A实现了91.4%(最高可达98.2%)的优异C1含氧产物选择性,显著优于其晶态类似物(TiO₂–ZrO₂–A800)以及大多数先前报道的光催化剂。结合结构、光谱和理论分析,该研究确立了这种非晶相优势的根源。非晶ZrO₂在破坏中程Zr–O–Zr连接性的同时保留了短程Zr–O配位,从而生成了丰富的Oᵥ和稳健的Ti–O–Zr界面连接。这些特征促进了电荷分离和传输,这体现在相较于A800略微变窄的光学带隙和更低的电荷转移电阻上,两者共同维持了更高的稳态空穴数量。重要的是,非晶界面还调节了ROS通量,在维持高效氧化的同时最大限度地减少了自由基驱动的过氧化。通过EPR自旋捕获和原位DRIFTS进行的机理研究支持了一个统一的反应路径,其中CH₄被光生h⁺和/或•OH活化形成•CH₃,随后与•OOH/•OH偶联/氧化生成CH₃OOH、CH₃OH和HCHO。通过缩短含氧中间体的表面停留时间并抑制过氧型深度氧化,非晶ZrO₂将反应网络导向液态含氧产物而非CO₂。该研究凸显了非晶相工程作为一种控制自由基微环境和反应路径的有效杠杆,为在温和条件下实现选择性CH₄官能团化提供了设计原则。


 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
