华南农业大学 Adv. Mater. | 引入缺电子单分子结:共价有机框架实现高效光催化合成过氧化氢
投稿 / 合作 / 安瓿瓶采购 / 加入 COFs & MOFs 交流群,请添加小编微信。微信号:COFsandH2O2
1. 研究背景及意义
过氧化氢作为一种环境友好且高效的氧化剂,在化学合成与环境修复等领域不可或缺。利用太阳能驱动双电子氧还原反应光催化合成过氧化氢,被认为是一种极具前景的绿色生产途径。共价有机框架因其结构可调和孔道有序的特点,在该领域展现出巨大潜力。然而,当前常规的富苯环共价有机框架面临着核心技术瓶颈:其骨架电子密度分布极其均匀,缺乏明显的缺电子位点,导致对氧分子的吸附和活化能力较弱;同时,光生载流子的随机迁移阻碍了电荷向催化活性位点的精准传递,严重限制了整体光催化效率。
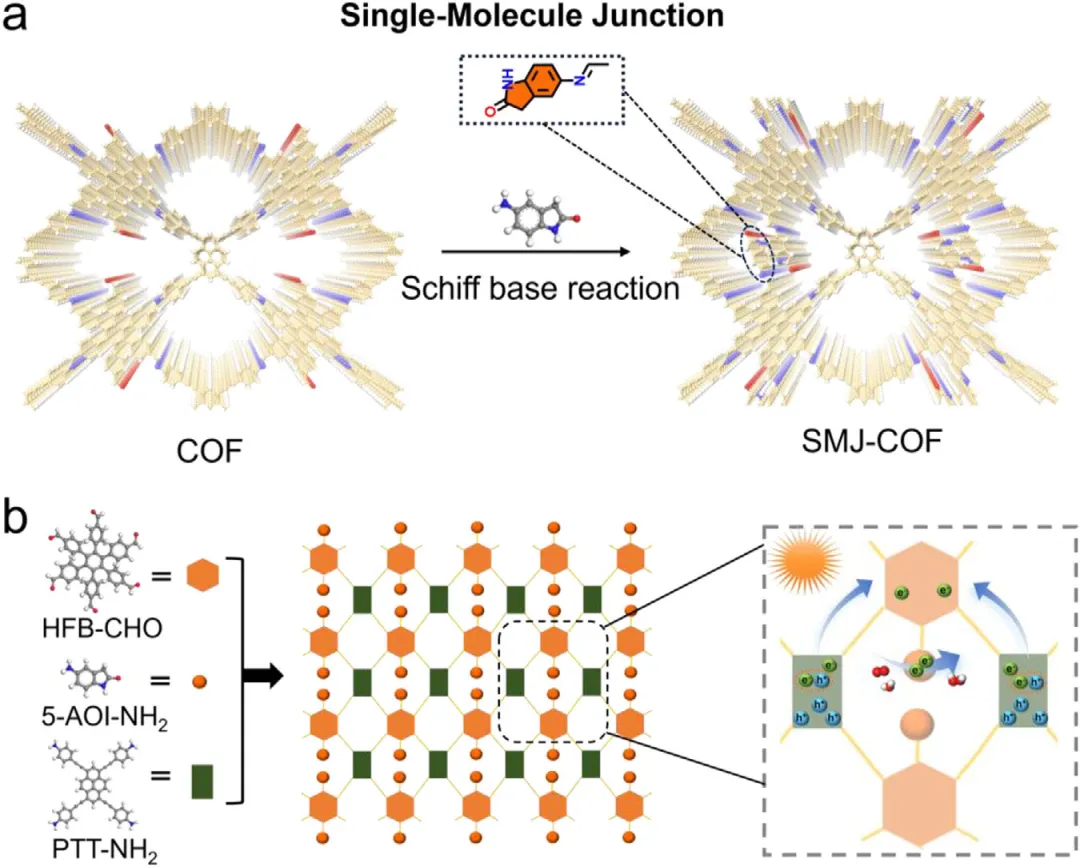
 为突破上述局限,本研究提出了一种利用单分子结策略精准构筑表面活性位点的创新设计思路。研究人员开发了一种通用的合成后修饰方法,通过一步席夫碱缩合反应,将具有局部缺电子特性的极性单分子结(如5-氨基羟吲哚)共价引入到亚化学计量比共价有机框架的未饱和位点和边缘残余位点中。
为突破上述局限,本研究提出了一种利用单分子结策略精准构筑表面活性位点的创新设计思路。研究人员开发了一种通用的合成后修饰方法,通过一步席夫碱缩合反应,将具有局部缺电子特性的极性单分子结(如5-氨基羟吲哚)共价引入到亚化学计量比共价有机框架的未饱和位点和边缘残余位点中。
该研究具有极其重要的方法学意义。它成功赋予了共价有机框架双重功能:局部缺电子中心能够强效锚定并活化氧分子,而单分子结的内置极性则为光生载流子构建了定向迁移通道,确保电子精准投递至反应位点。这一策略为克服有机半导体中电荷传输与底物活化的双重限制提供了极具普适性的分子工程范式。
2. 主要研究结果
2.1 实验方法
作者首先利用六醛基单体和四氨基单体合成了含有丰富残余基团的二维亚化学计量比共价有机框架前驱体。随后,采用一步后修饰策略,将5-氨基羟吲哚作为单分子结通过共价键嫁接到骨架上,构筑了目标材料SMJ-COF。与传统的从头合成或物理掺杂体系相比,该策略的新颖之处在于能够在完全不破坏材料本征晶体结构和连续多孔网络的前提下,精准且稳定地在孔道和边缘引入特定的活性中心。
2.2 研究亮点
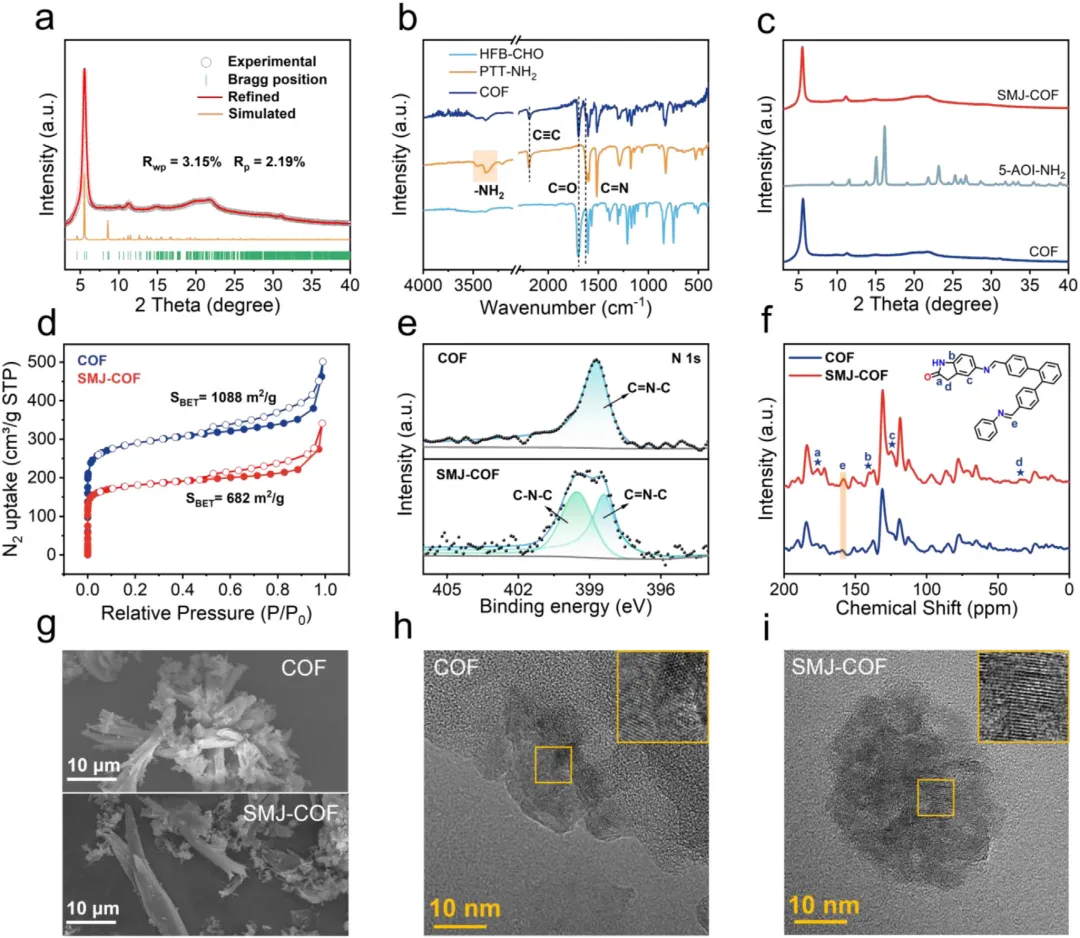
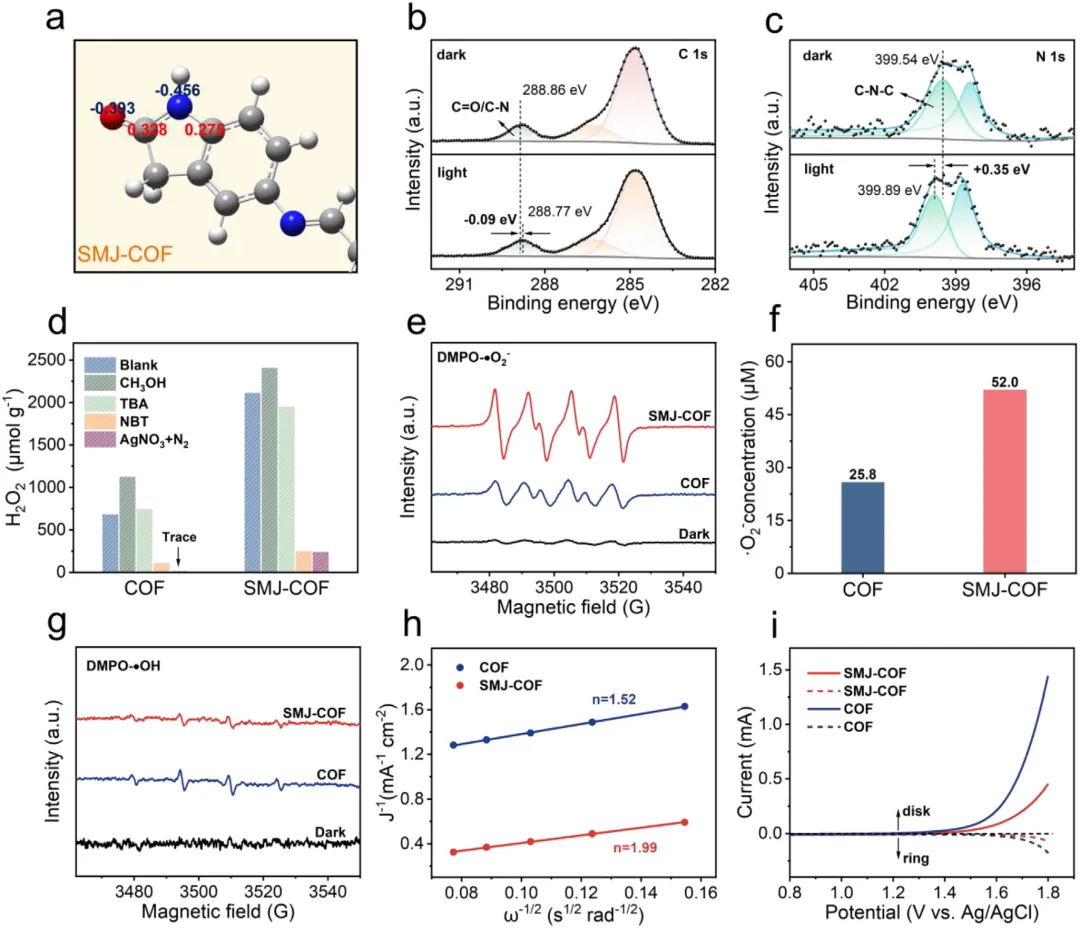
粉末X射线衍射证实修饰后的材料完美保持了原始的高结晶度,且未出现分子前驱体的游离峰。傅里叶变换红外光谱中五元环特征氮氢拉伸带的出现,以及固态碳13核磁共振与X射线光电子能谱中氮1s谱图分离出明确的碳-氮-碳及碳氮双键信号,确凿证明了单分子结被成功且共价地整合到骨架中。此外,氮气吸脱附测试显示比表面积从原始框架的1088 m2/g合理下降至682 m2/g,进一步证明了分子结成功占据并修饰了微观孔道。
2.3 核心性能表现
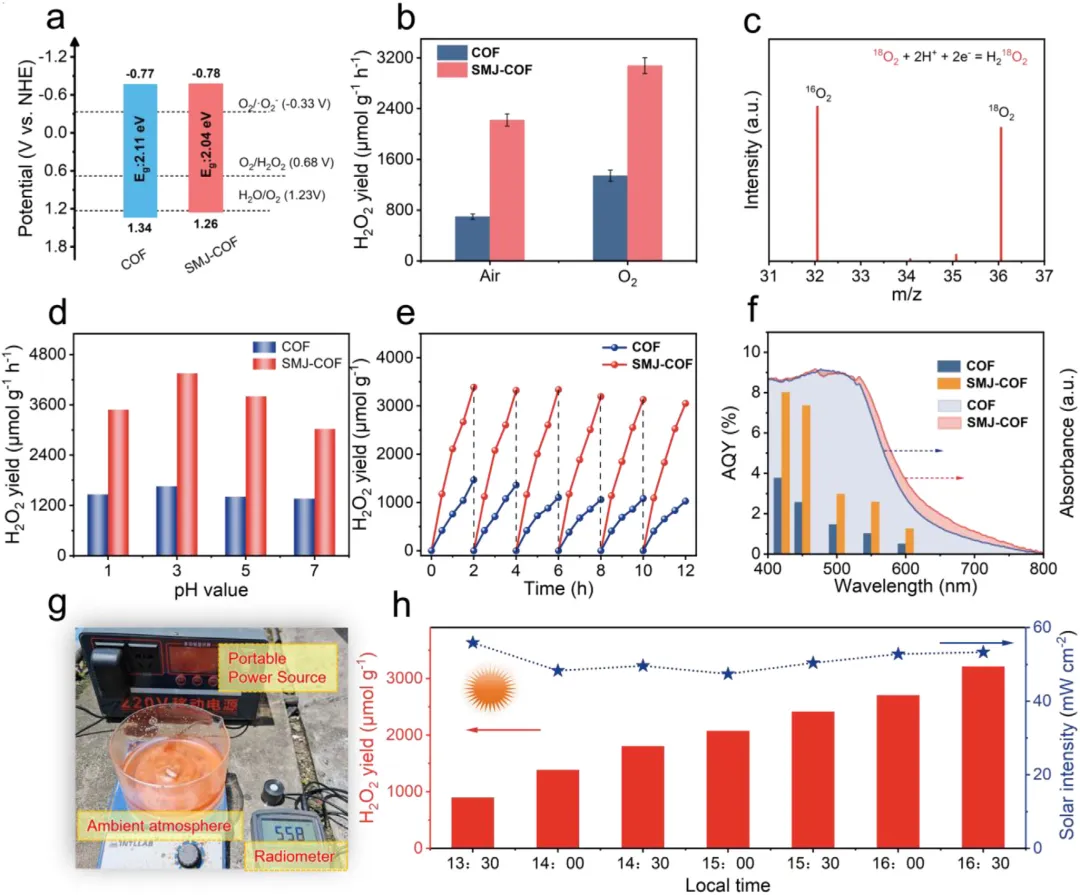
在可见光照射和酸性水溶液(pH值为3)条件下,优化后的SMJ-COF展现出4354 µmol/(g·h)的极高过氧化氢产率,是未修饰原始材料(1655 µmol/(g·h))的2.6倍。在纯水和空气的自然环境条件下,其产率依然高达2221 µmol/(g·h),达到原始材料的3.2倍。在420 nm处,该材料的表观量子产率达到8.02%,太阳能到化学能的转化效率高达0.49%。在循环稳定性方面,历经6次连续光催化测试后,其过氧化氢产率仍能保持初始性能的约90%,且微观形貌未发生明显变化。
2.4 机理验证
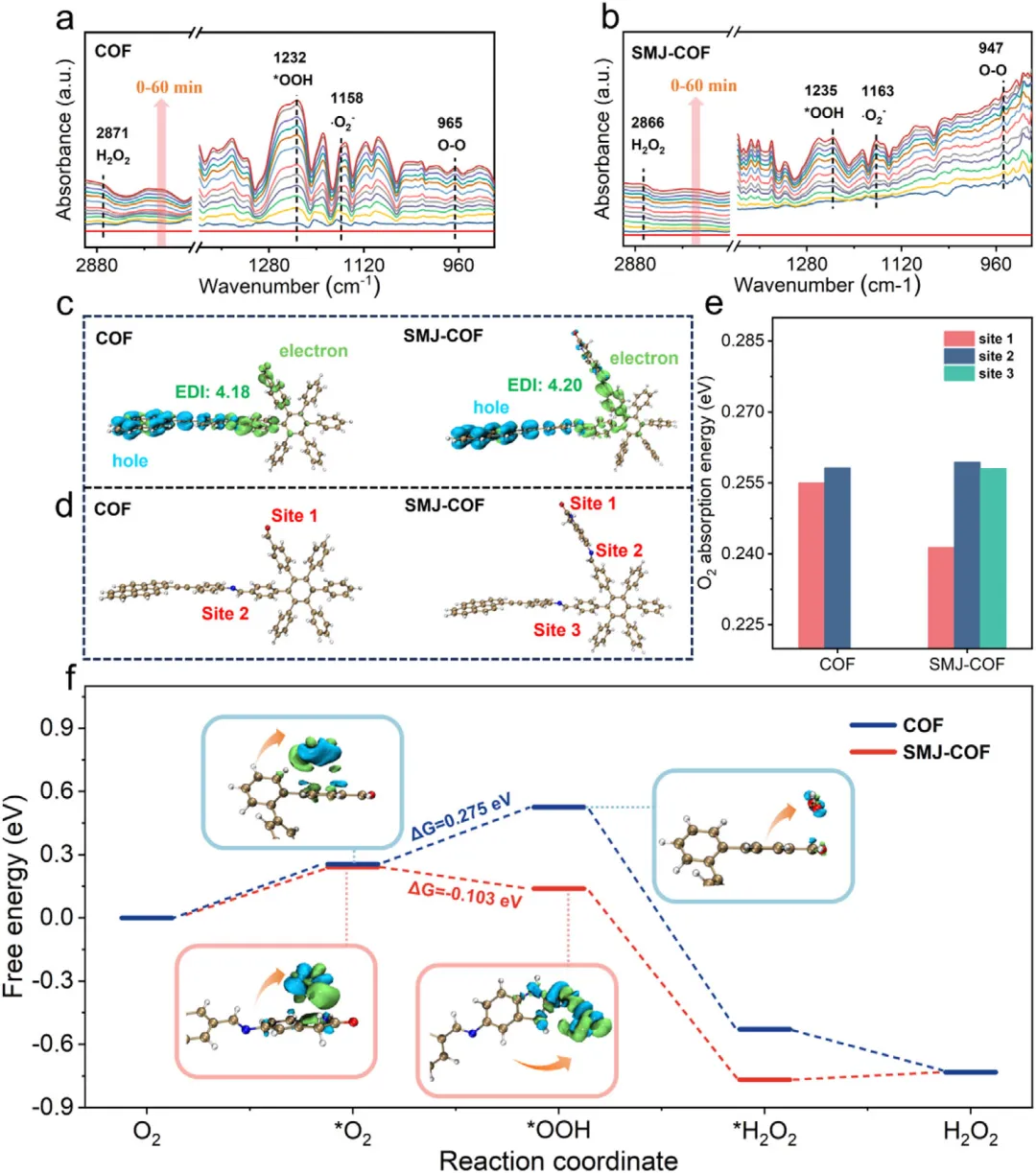
作者综合利用原位光谱、瞬态动力学测试与密度泛函理论计算深刻阐明了催化机理。在直接实验验证方面,氧18同位素标记实验确凿证明过氧化氢的氧原子来源于光催化氧还原过程。开尔文探针力显微镜和飞秒瞬态吸收光谱直接捕捉到SMJ-COF具有更显著的光诱导表面电势降(40.5 mV对比12.2 mV)以及更长的激发态寿命(18.17 ps对比10.36 ps),直接证明了单分子结极大促进了电荷的分离与定向传输。原位光照X射线光电子能谱观察到碳原子结合能的偏移,证实了光生电子在缺电子碳位点上的积累。基于以上证据结合密度泛函理论计算合理推断:单分子结的引入将体系的偶极矩从6.64 Debye提升至7.74 Debye,驱动了电荷的定向转移;同时,这一结构使决速步(超氧自由基转化为*OOH中间体)的吉布斯自由能从0.275 eV大幅降低至负0.103 eV,从热力学上将原本需要跨越能垒的过程转变为自发过程。
3. 研究结论
本研究成功开发了一种通过合成后修饰将局部缺电子极性分子嫁接至共价有机框架的通用策略,实现了在无牺牲剂条件下过氧化氢的高效光催化合成。这项工作的真正贡献在于打破了传统富苯环有机框架电子分布均匀导致底物活化弱的技术局限,提出了单分子结介导的“定向电荷投递”与“强化吸附活化”双重协同机制的新认识,为后续定制具有极化微环境和超高电荷利用率的先进光催化材料提供了极具启发性的通用平台。
https://doi.org/10.1002/adma.73233
本文使用科应智能助手(www.scienceing.com)开展文献调研

 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
