疟疾作为全球性的重大公共卫生问题,其病原体恶性疟原虫具备复杂的宿主免疫逃避策略。巨噬细胞在识别和清除疟原虫过程中发挥着极其关键的作用,但疟原虫具体如何调控感染红细胞逃避巨噬细胞识别一直是未解之谜。
2026年4月14日,沈阳农业大学陈启军教授团队在Immunity & Inflammation期刊上发表题为“Plasmodium PI3K suppresses the externalization of phosphatidylserine on infected erythrocytes”的最新研究成果。该研究深入揭示了恶性疟原虫通过其自身的磷酸肌醇3-激酶(PI3K)主动抑制感染红细胞表面磷脂酰丝氨酸外翻,从而规避宿主巨噬细胞监视与清除的分子机制。
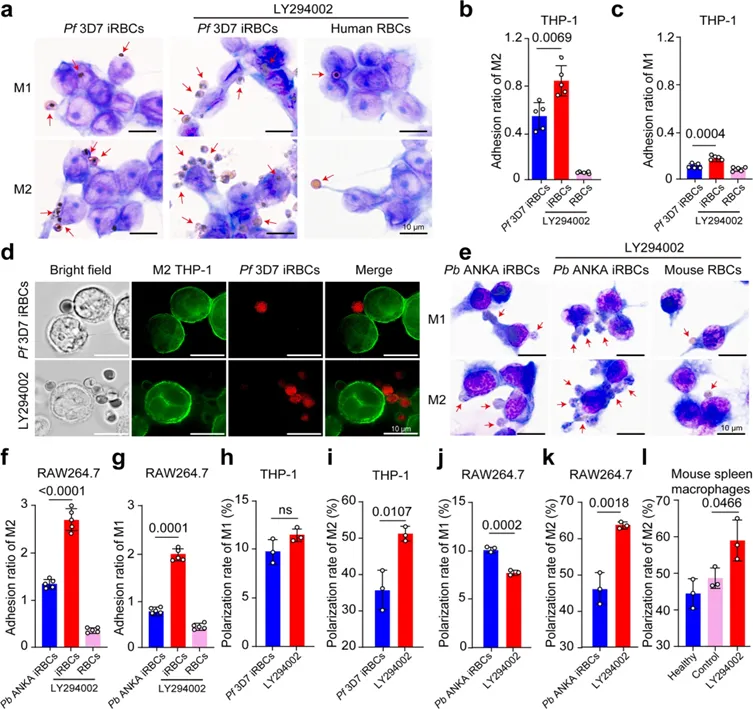
研究团队分别利用感染人的恶性疟原虫3D7虫株和感染鼠的伯氏疟原虫ANKA感染模型,系统阐释了虫源性PI3K在免疫逃逸过程中的核心功能。研究表明,疟原虫通过PI3K抑制磷脂酰丝氨酸在染虫红细胞表面的表达。当疟原虫PI3K的活性被抑制后,染虫红细胞表面的磷脂酰丝氨酸暴露水平出现显著上升。磷脂酰丝氨酸是细胞表面一种经典的促吞噬信号(“eat-me signal”),其大量外翻能够驱动宿主单核细胞向M2型巨噬细胞极化,并大幅提升M2型巨噬细胞对感染红细胞的识别、黏附与吞噬效率。这一现象在体内和体外实验中均得到了验证,说明疟原虫通过PI3K维持在宿主细胞内感染的隐蔽性(图1)。
图1 疟原虫通过PI3K维持在宿主细胞内感染的隐蔽性
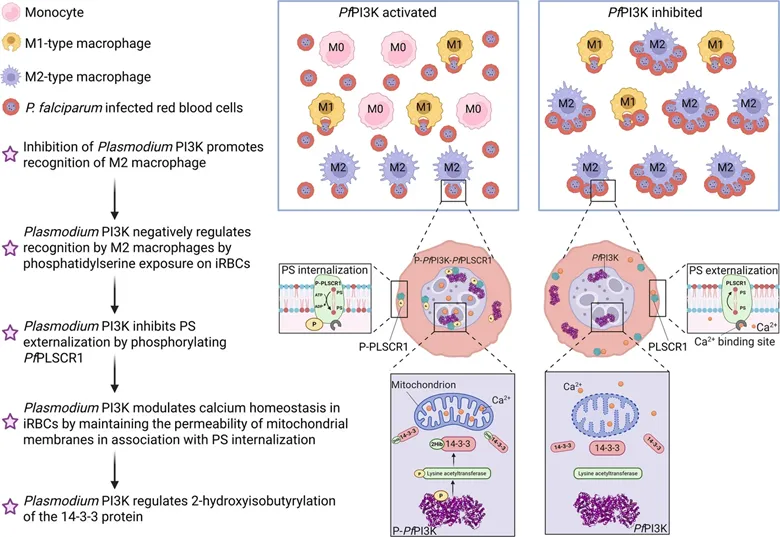
在分子机制层面,研究发现疟原虫PI3K通过调控两条相互关联的通路维持磷脂酰丝氨酸的内化状态。一方面,疟原虫PI3K可以通过直接磷酸化抑制疟原虫磷脂翻转酶1(PfPLSCR1)的活性,从源头上阻止了磷脂酰丝氨酸向细胞膜外侧的翻转。另一方面,疟原虫PI3K通过促进虫体线粒体14-3-3蛋白的2-羟基异丁酰化修饰,有效维持了虫体线粒体膜电位的稳定性,防止了细胞内钙离子的异常释放与外流。由于磷脂翻转酶的活性高度依赖于高浓度的钙离子,这种由PI3K调控的钙稳态环境进一步阻断了PfPLSCR1的活化通路(图2)。
图2 PfPI3K抑制染虫红细胞上磷脂酰丝氨酸外翻的机制示意图
该项研究系统地阐明了疟原虫利用特异性激酶网络调控宿主细胞膜脂质不对称分布的生理学机制。这一重要发现不仅丰富了对恶性疟原虫宿主-病原互作及免疫逃逸策略的科学认知,还揭示了一个极具价值的抗疟新靶点:通过靶向抑制疟原虫PI3K活性,促进宿主巨噬细胞对疟原虫寄生红细胞的识别和清除,为疟疾的临床干预提供了全新的理论基础。
原文链接:
https://link.springer.com/article/10.1007/s44466-026-00036-2
沈阳农业大学二级教授,博士生导师。现任中国畜牧兽医学会、中国动物学会副理事长。长期致力于人兽共患寄生虫病原生物学及致病机理研究,取得了一系列国际领先水平的原创性成果。
Immunity & Inflammation 是由曹雪涛院士和Jules A. Hoffmann 院士(2011年诺贝尔生理或医学奖获得者)担任主编,中国免疫学会主办,与国际出版商施普林格自然出版集团(Springer Nature)合作出版的开放获取英文期刊。Immunity & Inflammation 聚焦免疫与炎症领域重大科学问题和前沿进展,旨在促进免疫炎症相关基础研究与临床转化应用,收录方向包括但不限于免疫炎症机制、免疫炎症调控、炎症相关性疾病防治、免疫治疗、新技术新体系等。
诚邀来稿:
https://link.springer.com/journal/44466