云南农业大学农田无公害生产创新团队ACS Nano:氧化锰层状-隧道结构转化驱动重金属异常稳定封存
- 2026-06-21 11:03:40




第一作者:梁新然
通讯作者:梁新然、冯雄汉、万彪
通讯单位:云南农业大学资源与环境学院,华中农业大学资源与环境学院

图片摘要

成果简介
近日,云南农业大学资源与环境学院梁新然副教授联合国内外合作者,在国际权威Nature Index期刊ACS Nano发表题为“Anomalous Heavy Metal Sequestration via MnO2Layer-to-Tunnel Transformation”的研究论文。该研究围绕一个长期存在的环境科学问题展开:低结晶、强吸附能力的矿物在结晶化过程中,通常会因为比表面积下降和活性位点减少而释放已吸附的重金属;然而,本研究发现,层状δ-MnO2向2×2隧道结构α-MnO2转化时,不仅没有导致Pb2+大量释放,反而显著增强了其固定量和稳定性。研究表明,Pb2+首先与δ-MnO2表面的K+发生交换,随后在矿物定向组装和相变过程中进入新生成的α-MnO2隧道内部,实现由表面吸附向结构封存的转变,提出了矿物结晶化驱动重金属深度封存的新机制。

引言
在污染土壤与废水修复研究中,锰氧化物因其较高的比表面积、丰富的表面位点和较强的重金属吸附能力而被广泛关注。传统认识普遍认为,矿物由低结晶态向高结晶态转化时,往往伴随着已固定金属的再释放,尤其是由层状结构向隧道结构转化时,原有吸附位点减少,重金属更容易重新进入环境。然而,这一过程是否一定导致重金属失稳释放,仍缺乏直接而系统的证据。基于此,本研究以典型层状锰氧化物δ-MnO2为对象,结合XRD、XAFS、HRTEM、AC-HRTEM和DFT计算等多种手段,系统揭示了Pb2+在δ-MnO2向α-MnO2相变过程中的迁移、再固定及稳定化机制,并进一步讨论了不同尺寸金属离子在隧道结构中的差异化归趋。

图文导读
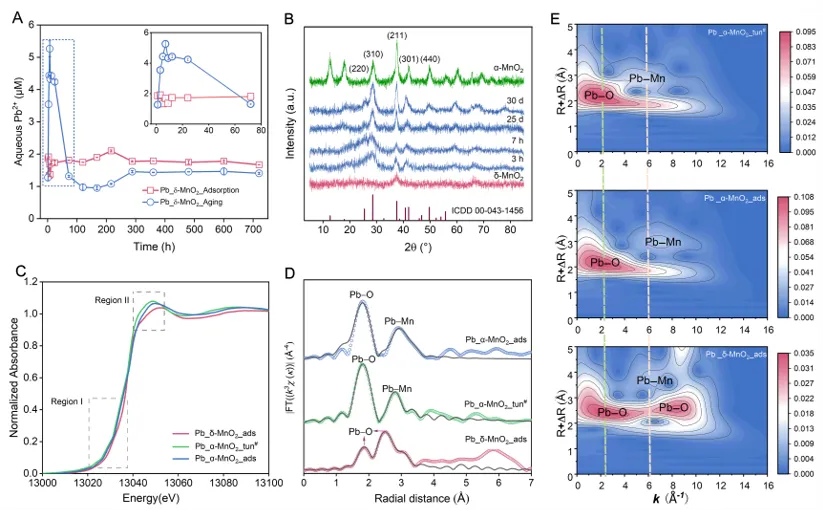
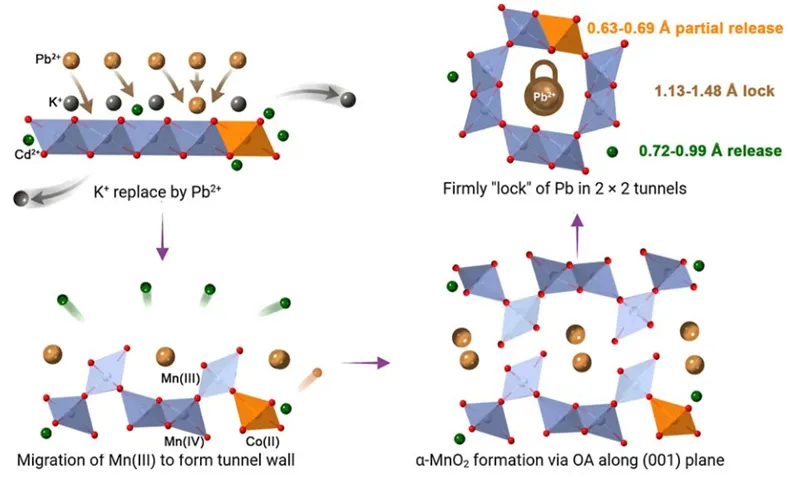
图1:Pb2+在pH 4条件下吸附于δ-MnO2并在Mn2+诱导下随矿物相变进入α-MnO2隧道结构的过程。
研究首先比较了Pb2+在δ-MnO2表面吸附,以及Mn2+诱导δ-MnO2老化/转化过程中溶液中Pb2+浓度的变化。结果显示,加入Mn2+后,Pb2+在最初阶段仅发生轻微释放,随后浓度持续下降,且在3-30 d内始终保持在极低水平。这说明在δ-MnO2向α-MnO2转化过程中,Pb并未因矿物结晶化而大量解吸。进一步的XRD结果表明,反应3 h后即出现α-MnO2特征衍射峰,并随时间推移逐渐增强,说明新相快速形成并不断提高结晶度。XANES/EXAFS结果则显示,Pb在转化产物中的配位环境更规则、Pb-Mn配位数更高,指向Pb并非停留于表面,而是进入了更稳定的结构位点。
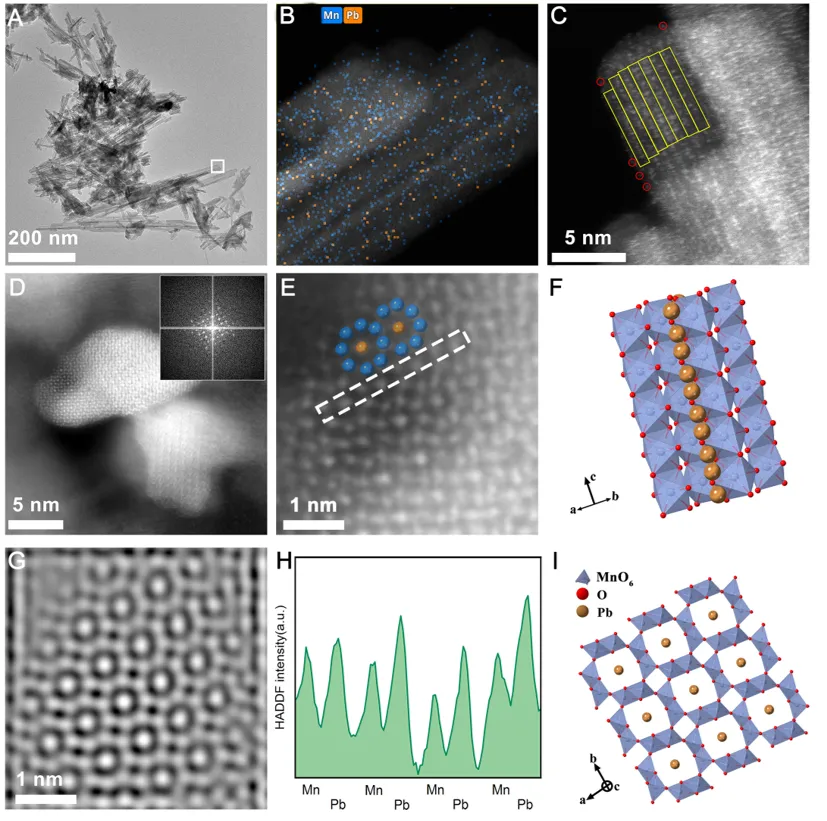
图2:AC-HRTEM等表征揭示Pb在新形成α-MnO2中的空间分布特征。
原子分辨成像进一步证实了上述判断。AC-HRTEM和HAADF-STEM结果显示,在新生成的α-MnO2纳米棒中,多个Pb原子沿隧道轴向有序排列,其原子间距与α-MnO2的晶面间距高度匹配,表明Pb是被封装于2×2隧道内部而非简单吸附在外表面。作者据此提出,Pb在δ-MnO2向α-MnO2转化过程中既充当隧道形成的“模板离子”,又作为结构稳定剂参与新相组装。也正因为如此,矿物相变不仅没有削弱Pb固定,反而构建出更稳定的结构性封存位点。
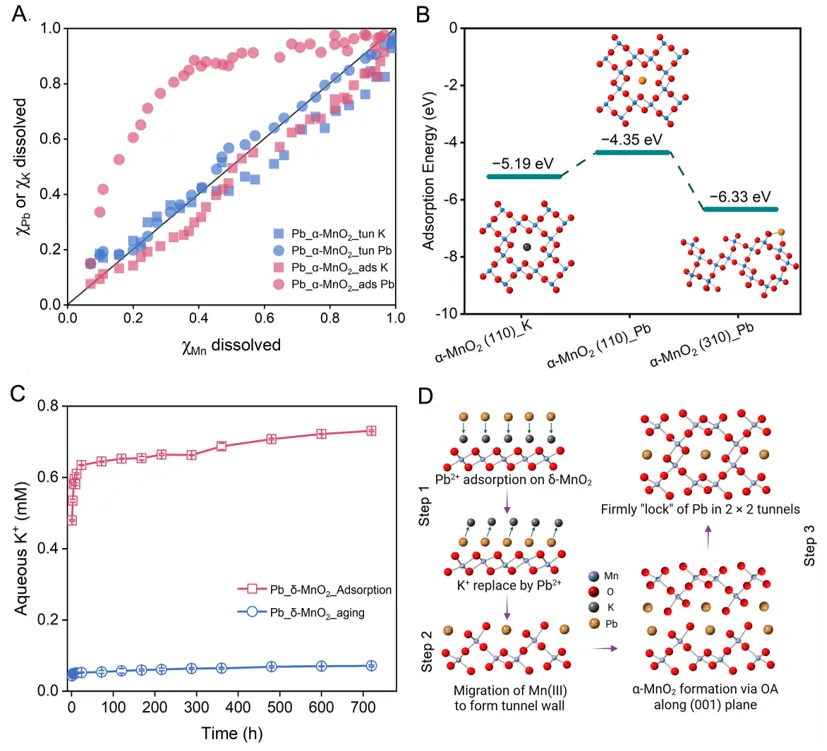
图3:酸溶实验、K+释放行为和DFT计算共同揭示Pb在不同位点上的稳定性差异。
为比较“表面吸附”和“隧道封存”两种结合方式的稳定性差别,研究进一步开展了酸溶实验。结果表明,隧道封存样品的Pb固定容量达到260.0 mg/g,约为合成α-MnO2表面吸附容量(22.3 mg/g)的12倍;完全溶解后释放出的Pb浓度也显著更高,说明转化生成的α-MnO2中实际容纳了更多Pb。与此同时,K+动态变化表明,Pb在吸附初期即可与δ-MnO2表面相关K+发生交换,而在后续相变过程中进一步替代隧道中的K+,最终被稳定“锁定”于α-MnO2内部。DFT计算同样支持这一结论:Pb位于隧道中的结合更加稳定,明显不同于外表面弱吸附状态。
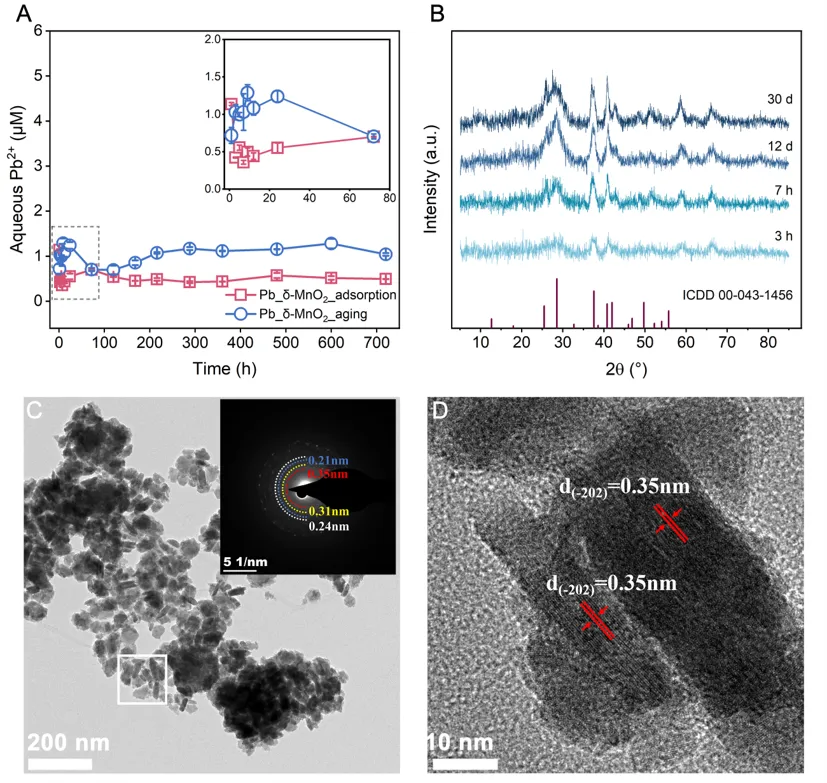
图4:pH 5.5条件下Pb吸附和Mn2+驱动δ-MnO2结构演化行为。
为检验该机制在不同环境条件下是否依然成立,作者在pH 5.5条件下进行了平行实验。结果显示,在这一条件下,单独加入Mn2+并不能促使δ-MnO2发生相变,但Pb的存在能够明显促进δ-MnO2向α-MnO2转化。反应结束时溶液中Pb2+浓度约为0.45 μM,甚至低于pH 4条件下的对应水平。HRTEM进一步观察到形成的α-MnO2纳米棒长度约为10-20 nm,说明较高pH会影响定向附着和晶体长大过程,但并不改变Pb随相变进入隧道并被稳定固定的总体机制。
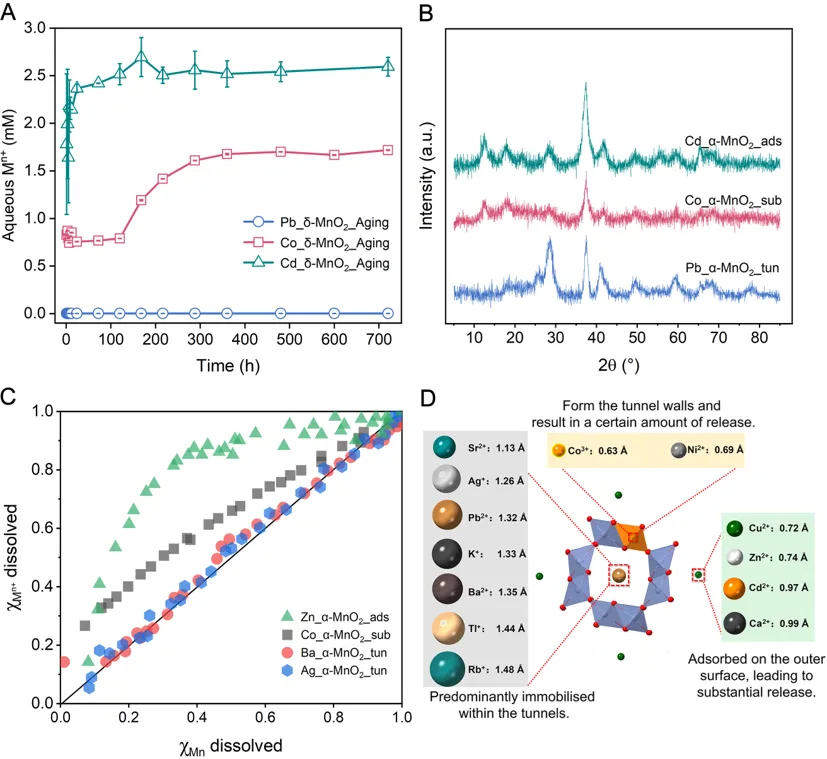
图5:不同尺寸金属离子在Mn2+诱导δ-MnO2向α-MnO2转化过程中的归趋差异。
研究还比较了多种金属离子的行为差异。结果表明,Pb2+、Ba2+和Ag+等与α-MnO2 2×2隧道尺寸较为匹配的离子,可在相变过程中进入隧道内部并形成更稳定的结构封存;Co2+等更小的离子更倾向于在隧道壁发生同晶置换;Cd2+和Zn2+则主要停留在表面吸附位点,难以进入隧道内部。基于这些结果,作者提出矿物结晶化诱导重金属稳定化主要受三个因素控制:一是离子尺寸与晶体空腔的匹配关系,二是目标离子对开放骨架的结构模板作用,三是矿物相变过程本身的拓扑演化路径。

小结
本研究提出并证实了一种不同于传统认识的重金属固定机制:在特定条件下,矿物由层状结构向隧道结构转化,不一定导致已吸附金属释放,反而可能通过定向组装和结构重构,实现对重金属的异常增强与持久封存。该工作不仅深化了对锰氧化物界面重金属地球化学行为的认识,也为污染土壤和废水中重金属的长期稳定化治理、矿物修复材料的结构设计以及选择性金属捕获策略的构建提供了新的理论依据。
该工作得到国家自然科学基金项目(42367005、42407326、42377303和W2521004)、国家自然科学基金优秀青年科学基金项目(海外)以及华中农业大学中央高校基本科研业务费专项资金资助。

作者简介

梁新然,云南农业大学资源与环境学院副教授,硕士生导师,云南省“兴滇英才支持计划”青年人才(引进)。2020年获得华中农业大学土壤学博士学位,2020年12月入职云南农业大学。研究方向为土壤矿物化学,聚焦于土壤铁锰氧化物晶体生长过程与有机质、重金属和其他环境因子互作机制。主持国家自然科学基金青年基金和地区项目各1项,在ACS Nano,Nature Communications,Environmental Science & Technology Letter,Environmental Science: Nano等期刊发表论文48篇。担任Proceedings of the National Academy of Sciences of the United States of America,Geoderma,Applied Geochemistry等国际知名期刊审稿人。

万彪,华中农业大学教授,博士生导师。2020年12月,美国佐治亚理工学院获博士学位。2021至2023年在德国图宾根大学完成博士后研究。“土壤过程与环境效应”院青年科研团队负责人,主要从事厌氧生物地球化学与厌氧微生物学研究,聚焦厌氧生境中微生物与矿物共同介导的铁-碳-磷多要素协同循环、微生物铁氧化还原、磷界面化学、产甲烷与甲烷氧化微生物机制、铁-氮或铁-碳协同转化等生物地球化学相关研究。主持国自然青年科学基金项目(C类)、国家青年人才项目、国家重点研发子课题、国家外国专家个人类项目(H类)等多项,曾主持美国、法国、英国同步辐射项目4项,参与美国科学基金(NSF)和德国科学基金会(DFG)项目。研究成果以第一作者或通讯作者(含共一、共通)发表在Cell Host Microbe、ACS Nano、Environ Sci Tech、Geochim Cosmochim Ac等国际权威期刊20余篇。担任Nature Geosci、Nat Commun、Environ Sci Tech等期刊审稿人。

冯雄汉,华中农业大学二级岗教授,博士生导师。主要从事土壤矿物化学研究,在土壤矿物结构与演化、界面反应过程与机理,及其生态环境效应等方面取得创新性成果。在国内外相关领域的高水平期刊ACS Nano, Environ Sci Technol, Geochim Cosmochim Ac, Water Res等国际著名期刊上以第一/通讯作者发表SCI收录论文85篇,包括Nature Index期刊论文23篇。在美国化学会出版英文专著《Advances in the Environmental Biogeochemistry of Manganese Oxides》1部。主持国家自然科学基金重点项目、面上项目和国际合作与交流项目,及国家重点研发计划课题等科研项目,担任国际期刊Journal of Soils and Sediments和Geochemical Transactions编委。曾获全国优秀博士论文奖,入选国家高层次人才特殊支持计划。
文章链接:https://doi.org/10.1021/acsnano.6c00961

投稿、合作、转载、进群,请添加小编微信Environmentor2020!环境人Environmentor是环境领域最大的学术公号,拥有25W+活跃读者。由于微信修改了推送规则,请大家将环境人Environmentor加为星标,或每次看完后点击页面下端的“在看”,这样可以第一时间收到我们每日的推文!环境人Environmentor现有综合群、期刊投稿群、基金申请群、留学申请群、各研究领域群等共20余个,欢迎大家加小编微信Environmentor2020,我们会尽快拉您进入对应的群。

往期推荐



扫描二维码,快速入群~
随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 四川农业大学四川省近五年录取数据(2025版:含录取人数、录取最低分、最低位次、学校排名、特色专业等)
- 农村发展研究主任和农业农村局科长,对比
- 农业农村部规划设计研究院齐飞研究员等:日光温室平椭圆管骨架套筒螺钉拼接抗弯性能
- 水利局局长和农业农村局局长,岗位价值
- 工业强区区长和农业大区区长,对比
- 【考研初试资料】2027年江西农业大学考研历年真题|大纲|参考书目|笔记|课件|复习提纲|题库|模拟卷
- 广东省农业农村厅党组理论学习中心组专题学习《信访工作条例》
- 陈石督导调研项目盘活、农业产业发展和森林防火工作
- 【智慧农业】小麦“一喷三防”技术指导意见
- 河南省农业农村厅关于开展2026年第二轮土地承包到期后再延长30年试点申报工作的通知
