在人工智能驱动药物发现的浪潮中,分子表示学习(MRL)已成为连接分子结构与功能预测的核心技术。传统的分子表示方法依赖人工设计的特征描述符,受限于人类总结的物理化学知识,难以全面捕捉分子的内在属性。近年来,基于自监督预训练的MRL方法取得显著进展,其中多模态预训练方法通过整合分子的一维序列、二维拓扑图、三维构象图等多源数据,能够学习到更丰富的分子表征。然而,现有方法在预训练阶段通常要求分子数据具备完整的模态信息,而真实场景中除了二维图结构外,其他模态数据往往缺失;同时,下游任务通常仅提供单一的二维分子图作为输入,导致多模态预训练模型的实用性受限。如何在不完整模态条件下实现多模态知识的高效迁移,成为该领域亟待解决的关键问题。
近日,华中农业大学章文教授团队联合美国俄亥俄州立大学张平教授在《Nature Communications》发表了题为“Multi-to-uni modal knowledge transfer pre-training for molecular representation learning”的研究论文。该论文于2026年正式接收发表,第一作者为熊展坤、王紫嫣、黄峰三位共同贡献者。研究团队提出了一种名为M2UMol的多模态知识迁移预训练框架,通过巧妙的“多对一”模态迁移策略,使模型能够在预训练阶段接受不完整的模态输入,并在仅提供二维分子图的下游任务中生成高质量的多模态分子表征。
研究团队首先构建了一个包含11571个药物样分子的多模态数据集,涵盖二维拓扑图、三维构象图、文本描述和生化特征四种模态。不同于传统方法仅能使用具备完整模态的4325个分子,M2UMol通过设计“生成-真实多模态对比学习”任务,可分别对齐二维分子生成的伪三维、文本、生化表征与对应的真实表征,从而使预训练数据规模扩大近三倍。同时,研究引入模态分类器作为辅助任务,确保生成的各模态表征能够保持模态特异性。这种设计使得M2UMol能够从11k规模的小型数据集中高效学习,在单张RTX 3090显卡上仅需11小时即可完成预训练。
在分子性质预测任务中,研究团队在8个基准数据集上采用更具挑战性的骨架划分(scaffold split)设置进行评估。结果显示,M2UMol在5个数据集上取得最佳性能,平均AUC提升幅度达5%至10%以上。值得注意的是,与需要百万级预训练数据的模型相比,M2UMol仅用11k分子就实现了更优的预测性能,展现出极高的数据效率和可扩展性。在分子相互作用预测任务中,M2UMol在药物-药物相互作用和药物-靶点相互作用任务上均表现优异。在冷启动和骨架划分两种挑战性场景下,M2UMol在DDI预测任务中相比基线方法在ACC指标上分别提升9.66%和6.84%;在DTI预测任务中,相较于DrugBAN方法,M2UMol在AUC和AUPR指标上平均提升2.60%和2.48%。
为验证多模态知识迁移的有效性,研究团队对生成的伪多模态表征质量进行了系统评估。通过t-SNE可视化发现,生成的三维、文本、生化表征能够与真实表征形成清晰的模态聚类,表明模型成功捕获了不同模态的差异化知识。在多模态检索任务中,生成的三维表征质量最高(Recall@1=0.82),文本表征次之(Recall@1=0.72),生化特征由于二值向量的稀疏性相对较低(Recall@1=0.35),但仍达到可接受水平。以抗炎药物二氟拉松为查询分子的案例研究表明,在不同模态表征空间中检索到的邻近分子具有与查询分子相似的结构特征、药理作用或靶点信息,证实模型能够捕捉分子间的内在关联。
研究进一步通过注意力权重可视化揭示了M2UMol识别分子关键结构的能力。以羟甲唑啉为例,当预训练模态从二维逐步增加到全部四种模态时,模型的注意力逐渐聚焦于羟基和咪唑啉环——这两个基团正是该药物与肾上腺素能受体结合时参与氢键相互作用和离子相互作用的关键活性基团。对比实验表明,纳入的模态越多,模型对分子关键结构的识别越精准。这一发现证实M2UMol能够仅基于多模态分子数据本身理解分子的构效关系,无需依赖标注信息。
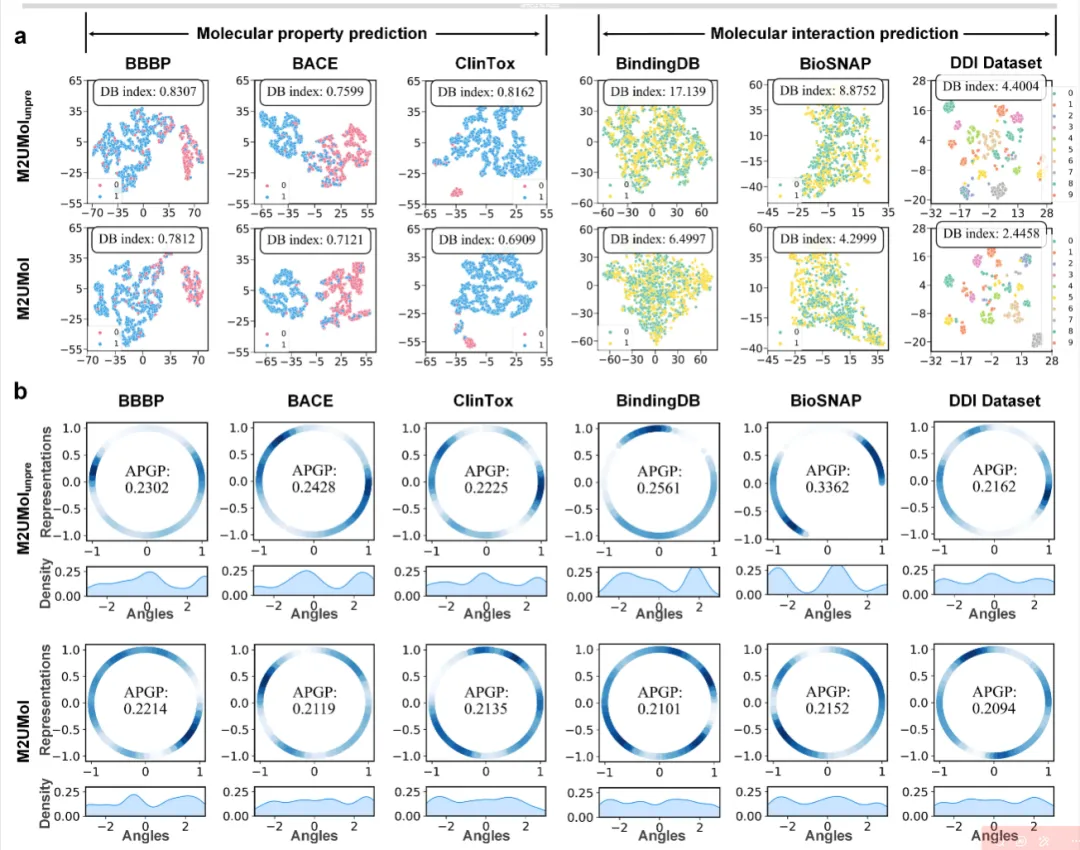
在下游任务表征质量分析中,研究对比了经过预训练的M2UMol与未经预训练的版本在测试集上的表征分布。结果显示,M2UMol学习到的正负样本表征在BBBP和BACE数据集上分离更加清晰;在DTI任务中DB指数显著降低;在DDI任务中同类样本聚类更加紧密。通过高斯核密度估计和平均成对高斯势(APGP)分数评估,M2UMol生成的分子表征在单位超球面上分布更为均匀,APGP分数更低,角度密度估计曲线更加平滑,表明模型能够学习到包含更丰富信息的分子表征,避免表征坍缩问题。
在模型可解释性分析中,研究对氟尿嘧啶、苯肼、阿司匹林等分子的注意力权重进行可视化。以氟尿嘧啶为例,M2UMol重点关注其卤素基团和两个酰胺基团——前者是该分子产生毒性的关键基团,后者与其可能引起高氨血症的副作用相关。在DDI预测任务中,以洛索洛芬为查询分子,模型准确识别出其丙酸基团中的羧基为关键活性基团,并在一同服用的利尿药和降压药中准确定位与药物相互作用相关的功能基团,如呋塞米的磺酰基和氨基、肼屈嗪的肼基等。这些可视化结果与人类对药物分子结构特性的认知高度一致。
基于M2UMol框架,研究团队开发了用户友好的软件包,集成了分子表示学习、多模态数据检索、关键结构识别等功能。该研究成果为药物发现领域的AI应用提供了新的技术路径,其代码、预训练权重和软件包已在GitHub平台开源发布。
欢迎关注本公众号,所有内容欢迎点赞👍,推荐❤️,评论,转发~
文末点击左下角阅读原文,可跳转文献原文链接~~